(12) Oversettelse av europeisk patentskrift
|
|
|
- Konrad Sørensen
- 8 år siden
- Visninger:
Transkript
1 (12) Oversettelse av europeisk patentskrift (11) NO/EP B1 (19) NO NORGE (1) Int Cl. C07K 16/42 ( ) A01K 67/027 ( ) A61P 37/06 ( ) Patentstyret (21) Oversettelse publisert (80) Dato for Den Europeiske Patentmyndighets publisering av det meddelte patentet (86) Europeisk søknadsnr (86) Europeisk innleveringsdag (87) Den europeiske søknadens Publiseringsdato (30) Prioritet , US, P (84) Utpekte stater AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MT NL NO PL PT RO SE SI SK TR Utpekte samarbeidende stater AL BA MK RS (73) Innehaver Genentech, Inc., 1 DNA Way, South San Francisco, CA 94080, US-USA (72) Oppfinner WU, Lawren, 1 Pensacola Street, Foster City, California 94404, US-USA BALAZS, Mercedesz, 273 Driftwood Street, Hayward, California 944, US-USA BRIGHTBILL, Hans, 240 Dolores Street, 124, San Francisco, California 943, US- USA CHAN, Andrew, 1201 Cloud Avenue, Menlo Park, California 9402, US-USA CHEN, Yvonne, 222 8th Avenue, 320, San Mateo, California 94401, US-USA CHUNTHARAPAI, Anan, 826 Ellis Drive, Colma, California 9401, US-USA DENNIS, Mark, 120 Plymouth Avenue, San Carlos, California 94070, US-USA WONG, Terence, 312 Shell Gate Road, Alameda, California 9401, US-USA (74) Fullmektig Zacco Norway AS, Postboks 2003 Vika, 012 OSLO, Norge (4) Benevnelse APOPTOTISKE ANTISTOFFER MOT IGE SOM BINDER MEMBRANBUNDET IGE (6) Anførte publikasjoner CHEN HUAN YUAN ET AL: "Monoclonal antibodies against the C(epsilon)mX domain of human membrane-bound IgE and their potential use for targeting IgE-expressing B cells." INTERNATIONAL ARCHIVES OF ALLERGY AND IMMUNOLOGY AUG 2002, vol. 128, no. 4, August 2002 ( ), pages , XP ISSN: CHANG TSE WEN: "Developing antibodies for targeting immunoglobulin and membrane-bound immunoglobulin E." ALLERGY AND ASTHMA PROCEEDINGS : THE OFFICIAL JOURNAL OF REGIONAL AND STATE ALLERGY SOCIETIES 2006 MAR-APR, vol. 27, no. 2 Suppl 1, March 2006 ( ), pages S7-14, XP ISSN: CHEN HUAN YUAN ET AL: "Generation and characterization of monoclonal antibodies against a segment (epsilonm67) uniquely present in membrane-bound IgE" FASEB JOURNAL, vol. 1, no., 8 March 2001 ( ), page A18, XP & ANNUAL MEETING OF THE FEDERATION OF AMERICAN SOCIETIES FOR EXPERIMENTAL BIOLOGY ON EXPERIMENTAL BIOL; ORLANDO, FLORIDA, USA; MARCH 31-APRIL 04, 2001 ISSN: POGGIANELLA MONICA ET AL: "The extracellular membrane-proximal domain of human
 Den europeiske søknadens Publiseringsdato 2009.12.16 (30) Prioritet 2007.03.")
2 membrane IgE controls apoptotic signaling of the B cell receptor in the mature B cell line A20." JOURNAL OF IMMUNOLOGY (BALTIMORE, MD. : 190) 1 SEP 2006, vol. 177, no. 6, 1 September 2006 ( ), pages , XP ISSN: TUMAS D B ET AL: "Anti-IgE efficacy in murine asthma models is dependent on the method of allergen sensitization." THE JOURNAL OF ALLERGY AND CLINICAL IMMUNOLOGY JUN 2001, vol. 7, no. 6, June 2001 ( ), pages 2-33, XP ISSN: INFÜHR D ET AL: "Molecular and cellular targets of anti-ige antibodies." ALLERGY AUG 200, vol. 60, no. 8, August 200 (200-08), pages , XP ISSN: FEICHTNER STEFAN ET AL: "Targeting the extracellular membrane-proximal domain of membrane-bound IgE by passive immunization blocks IgE synthesis in vivo." JOURNAL OF IMMUNOLOGY (BALTIMORE, MD. : 190) 1 APR 2008, vol. 180, no. 8, 1 April 2008 ( ), pages 499-0, XP ISSN:
, pages 2-33, XP002494042 ISSN: 0091-6749 INFÜHR D ET AL: \"Molecular and cellular targets of anti-ige antibodies.\" ALLERGY AUG 200, vol. 60, no.")
3 1 APOPTOTISKE ANTISTOFFER MOT IGE SOM BINDER MEMBRANBUNDET IGE RELASJON TILBAKE I TID Etter 3 USC 119 (e) krever oppfinnelsen prioritet over USSN 60/896,339, arkivert den 22. mars OPPFINNELSENS BAKGRUNN OPPFINNELSENS OMRÅDE 1 Oppfinnelsen vedrører apoptotiske antistoffer mot IgE, nukleinsyrer som koder for disse, terapeutiske sammensetninger som omfatter disse og anvendelse av dem ved behandling av IgE-formidlede forstyrrelser. BESKRIVELSE AV DEN BESLEKTEDE TEKNIKKEN Allergi står for visse sykdommer der immunresponsen mot miljømessige antigener forårsaker vevsinflammasjon og organdysfunksjon. De kliniske egenskapene til hver av de allergiske sykdommene gjenspeiler den immunologiske inflammasjonsreaksjonen i det involverte organet eller vevet. Disse egenskapene er generelt uavhengige av de kjemiske eller fysiske egenskapene til antigenet. Mangfoldet av allergiske reaksjoner oppstår når ulike immunologiske virkningsbaner involveres, og hver av dem danner et unikt inflammasjonsmønster. Allergi er vanlig over hele verden. Tilbøyeligheten for bestemte sykdommer vil imidlertid variere mellom ulike aldersgrupper, kjønn og raser. Forekomsten av følsomhet for spesifikke allergener bestemmes både av genetiske tilbøyeligheter og av geografiske og kulturelle faktorer som er ansvarlig for eksponeringen for allergenet. En klinisk allergitilstand påvirker bare noen individer som treffer på hvert allergen. Opptreden av allergisk sykdom etter eksponering for et allergen krever ikke bare forutgående «sensibilisering», men også andre faktorer som bestemmer lokaliseringen av reaksjonen til et bestemt organ. 3 En biologisk prosess som går forut for allergisykdommen etter allergen- eksponeringen gir opphav til en immunrespons som kalles «sensibilisering» eller sensibiliseringsfase. Når sensibiliseringen oppstår, blir ikke en person

4 2 symptomatisk før neste eksponering for allergenet. Sensibiliseringsvirkningen er også kjent som immunminne En av de primære reaksjonsveiene som fører til inflammasjon går gjennom immunoglobulin E (IgE). IgE spiller en sentral rolle i allergier på grunn av rollen sin som allergenreseptor på overflaten til mastceller og basofiler. IgEantistoffer er festet til overflaten av mastceller og basofiler i Fc-delen av molekylet til en celleoverflatereseptor med høy affinitet, omtalt som FcεRI. Den allergiske reaksjonen starter når det flerverdige allergenmolekylet bindes til antistoffer som opptar disse reseptorene. Resultatet er en brodannelse i FcεRI, som i sin tur signalerer intracellulært og fører til frigjøring og aktivering av formidlere av inflammasjon: histamin, leukotriener, kjemotaktiske faktorer, blodplateaktiverende faktor og proteinaser. Disse aktiverte formidlerne virker lokalt og fører til økt vaskulær permeabilitet, vasodilatasjon, sammentrekking av den glatte muskulaturen og sekretutskilling fra slimkjertler. Slike aktiviteter betegnes klinisk som den umiddelbare eller tidlige fasen, og opptrer i løpet av de første 1 30 minuttene etter allergeneksponeringen. I løpet av de neste 12 timene oppstår det progressiv vevsinfiltrasjon av inflammasjonsceller, fra nøytrofiler til eosinofiler og videre til mononukleære celler som respons på andre kjemiske formidlere som vi ikke forstår fullt ut. Denne tidsperioden på 6 12 timer etter allergeneksponeringer kalles den sene fasen og er karakterisert ved kliniske manifestasjoner av celleinflammasjon. Siden senfasereaksjonene, særlig i lungene, opptrer uten tidligfasereaksjoner, forstår vi fortsatt ikke helt om senfasereaksjonen nødvendigvis er IgE-formidlet. IgE eksisterer i en membranbundet form og i en utskilt form. Disse forskjellige formene synes å være spleisevarianter. Tidligere tilnærminger for å oppnå terapeutisk virkning ved å nedregulere IgE rettet seg hovedsakelig mot den utskilte formen (f.eks. XOLAIR omalizumab), for å hindre eller desarmere ytterligere «armering» av immunsystemet. Den utskilte formen av IgE er en kortere form, i det vesentlige slutter Fc-regionen ved CH4-domenet (figur 1), mens den lengre formen har flere rester i C-enden, inkludert peptidene som kodes for av eksonene som kalles M1/M1' og M2. Noen har rapportert to distinkte former av membranbundet IgE, både med og uten et 2-aminosyresegment som kalles M1' [Batista et al., J. Exp. Med. 184: (1996)], men søkerne kunne ikke bekrefte at noen membranbundet form mangler dette M1'-segmentet. Konvensjonell behandling med antistoffer mot IgE, som binder den utskilte formen

5 3 av IgE reduserer det frie serum-ige, men ikke det totale serum-ige. Casale et al., J. Allergy Clin. Immunol. 0 (1): (1997). 1 Chen et al. (2002) International Archives of Allergy and Immunology 128(4), , Chang (2006) Allergy and Asthma Proceedings: The Official Journal of Regional and State Allergy Societies 27(2), s. 7-14, Chen et al. (2001) Faseb Journal 1(), A18 og Poggianella et al. (2006) Journal of Immunology 177(6), dreier seg generelt om M1'-segmentet av IgE. Chang (2006) beskriver domenet på 2 aminosyrer mellom CH4-domenet og det C- terminale membranforankrede peptidet av IgE, kalt epsilon-domenet. Hver for seg beskriver Chen et al. (2002) og Chen et al. (2001) antistoffer som binder dette domenet, generelt eller ved en region i nærheten av C-enden. Separat rapporterer Poggianella et al. (2006) at dette domenet, kalt det ekstracellulære membranproksimale domenet (EMPD), kan være involvert i apoptose. Det har videre blitt observert at i fravær av antigensignal, er B-cellereseptorer (dvs. immunoglobuliner) som har tverrbindinger, utsatt for apoptose. 20 Søkerne har overraskende funnet at hvis det rettes IgE-antistoffer mot en region i N-enden av M1 -segmentet kan det utløse apoptose i B-cellen. Ettersom avkommet av aktiverte B-celler kan gi opphav til plasmaceller som lager og skiller ut den utskilte formen av IgE, gir desimering av IgE-produserende B- celler gjennom apoptose en ny terapeutisk tilnærming til allergibehandlingen. 2 OPPSUMMERING AV OPPFINNELSEN 30 Den foreliggende oppfinnelsen tilveiebringer apoptotiske antistoffer mot IgE, eller funksjonelle fragmenter av disse, og anvendelse av dem ved behandling av IgE-formidlede forstyrrelser som definert i kravene. Oppfinnelsen tilveiebringer også sammensetninger, fremgangsmåter for å hemme produksjon og utskilling av IgE fra B-celler og fremgangsmåter for å desimere IgE-produserende B-celler spesifikt, samt å senke det totale serum-ige som definert i kravene. 3 I en utførelsesform tilveiebringer oppfinnelsen et antistoff mot IgE/M1' som binder M1'-segmentet av IgE spesifikt og som utløser apoptose i IgE-uttrykkende B-celler som definert i kravene. I et spesifikt aspekt desimerer antistoffet spesifikt IgEproduserende B-celler. I et annet spesifikt aspekt, senker antistoffet det total
 beskriver domenet på 2 aminosyrer mellom CH4-domenet og det C- terminale membranforankrede peptidet av IgE, kalt epsilon-domenet. Hver for seg beskriver Chen et al. (2002) og Chen et al.")
6 4 serum-ige. I nok et annet spesifikt aspekt, senker antistoffet både totalt og fritt serum-ige. I et ytterligere spesifikt aspekt er serum-ige allergenspesifikt. I enda et ytterligere spesifikt aspekt, binder antistoffet IgE som stammer fra menneske, rhesusape og krabbemakak. I enda et ytterligere spesifikt aspekt er antistoffet kimært. I enda et ytterligere spesifikt aspekt er antistoffet humanisert. I enda et ytterligere spesifikt aspekt er antistoffet humant. 1 I en annen utførelsesform tilveiebringer den foreliggende oppfinnelsen et antistoff mot IgE/M1' som spesifikt binder en hvilken som helst av M1'- epitopene som svarer til peptidene som er identifisert i figur. I et spesifikt aspekt binder antistoffet spesifikt den samme epitopen som bindes av et antistoff valgt fra gruppen som består av: 47H4, 7A6, 26A11, 47H4v, 7A6v1 og 26A11v6. I et annet spesifikt aspekt binder antistoffet en epitop som svarer til et peptid valgt fra gruppen som består av: peptid 4 (SEQ ID NO:8), peptid (SEQ ID NO:9), peptid 7 (SEQ ID NO:11) eller peptid 8 (SEQ ID NO:12). I nok et annet spesifikt aspekt, binder antistoffet peptid 4 (SEQ ID NO:8), 20 I nok en annen utførelsesform tilveiebringer oppfinnelsen en M1'-epitop av IgE valgt fra gruppen som består av: peptid 4 (SEQ ID NO:8), peptid (SEQ ID NO:9), peptid 7 (SEQ ID NO:11) og peptid 8 (SEQ ID NO:12) som definert i kravene. I et spesifikt aspekt er MI'-peptidet peptid 4 (SEQ ID NO:8) I nok en ytterligere utførelsesform tilveiebringer oppfinnelsen et antistoff mot IgE som spesifikt binder et M1'-segment av IgE med en Scatchard-bindingsaffinitet til human IgE som er likeverdig med bindingsaffiniteten til det murine antistoffet mot IgE/M1, 47H4, eller den humaniserte varianten av dette som definert i kravene. I et spesifikt aspekt er affiniteten likeverdig med bindingsaffiniteten til 47H4. I et annet spesifikt aspekt er affiniteten mellom 0,30 og 0,83 nm. I nok et annet spesifikt aspekt er affiniteten likeverdig med bindingsaffiniteten til 47H4v. I et ytterligere spesifikt aspekt er affiniteten rundt 1, nm. 3 I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen et antistoff mot IgE/M1' som omfatter HVR i den tunge og den lette kjeden til antistoffet som fremkommer i en hvilken som helst av figurene 6A 6F som definert i kravene. I et spesifikt aspekt omfatter antistoffet også de variable regionene til den tunge og den lette kjeden til antistoffsekvensene som forekommer i en hvilken som helst av figurene 6A 6F. I et annet spesifikt aspekt omfatter antistoffet den fulle lengden av

7 den tunge og lette kjeden til antistoffsekvensene som fremkommer i en hvilken som helst av figurene 6A 6F. I nok et annet spesifikt aspekt fremkommer den tunge og den lette kjeden til antistoffsekvensene i en hvilken som helst av figurene 6A 6F. I et ytterligere spesifikt aspekt velges antistoffet fra gruppen som består av: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. I enda et ytterligere spesifikt aspekt er antistoffet 47H4v. I enda et ytterligere spesifikt aspekt er antistoffet afukosylert I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen en sammensetning som omfatter et antistoff mot IgE/M1' som omfatter HVR i den tunge og den lette kjeden til antistoffet som fremkommer i en hvilken som helst av figurene 6A 6F i kombinasjon med minst én farmasøytisk aksepterbar bærer som definert i kravene. I et spesifikt aspekt velges antistoffet fra gruppen som består av: 26A11, 26A11v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. I et annet spesifikt aspekt er antistoffet 47H4v. I nok et annet spesifikt aspekt er antistoffet afukosylert. I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen en sammensetning som omfatter et antistoff mot IgE/M1' som omfatter HVR i den tunge og den lette kjeden av antistoffet som fremkommer i en hvilken som helst av figur 8 13 i kombinasjon med ett eller flere legemidler valgt fra gruppen som består av: antistoff mot IgE, antihistamin, bronkodilator, glukokortikoid, NSAID, TNF-antagonist, integrinantagonist, immunundertrykkende middel, IL-4-antagonist, IL-13-antagonist, dobbel IL- 4/IL-13-antagonist, DMARD, antistoff som binder til en B-celleoverflatemarkør, og BAFF-antagonist som definert i kravene. I et spesifikt aspekt omfatter sammensetningen videre minst én farmasøytisk aksepterbar bærer I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen isolert nukleinsyre som koder for HVR i den tunge kjeden til antistoff mot IgE/M1' som fremkommer i en hvilken som helst av figurene 6A 6F som definert i kravene. I et spesifikt aspekt omfatter den isolerte nukleinsyren nukleinsyre som koder for HVR i den lette kjeden til antistoffet mot IgE/M1' som fremkommer i en hvilken som helst av figurene 6A 6F. I et annet spesifikt aspekt er antistoffet kimært. I nok et annet spesifikt aspekt er antistoffet humanisert. I et ytterligere aspekt er antistoffet humant. I nok et ytterligere spesifikt aspekt velges antistoffet fra gruppen som består av: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. I enda et ytterligere spesifikt aspekt er antistoffet 47H4v. I enda et ytterligere spesifikt aspekt er antistoffet afukosylert. I enda et ytterligere aspekt omfatter

8 6 nukleinsyren også en vektor som er egnet for uttrykking av nukleinsyren som definert i kravene. I enda et ytterligere spesifikt aspekt omfatter vektoren også en vertscelle som er egnet for uttrykking av nukleinsyrene som definert i kravene. I enda et ytterligere spesifikt aspekt er vertscellen en eukaryotisk celle eller en prokaryotisk celle. I enda et ytterligere spesifikt aspekt er den eukaryotiske cellen en pattedyrcelle, som f.eks. eggstokkceller fra kinesisk dverghamster (CHO). I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen en prosess for å lage et antistoff mot IgE/M1' eller funksjonelt fragment av dette, som spesifikt binder et M1'-segment av IgE, som omfatter å dyrke en vertscelle som inneholder nukleinsyre som koder for dette antistoffet eller fragmentet i en form som er egnet for uttrykking, under forhold som er egnet til å produsere et slikt antistoff eller fragment, og utvinne antistoffet eller fragmentet som definert i kravene I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen en produsert gjenstand som omfatter en beholder som omslutter en sammensetning som offentliggjøres i dette dokumentet og et pakningsvedlegg som indikerer anvendelse til å behandle en IgE-formidlet forstyrrelse som definert i kravene. I et spesifikt aspekt er den produserte gjenstanden et glass. I nok et annet spesifikt aspekt er den produserte gjenstanden en forhåndfylt sprøyte. I nok et annet spesifikt aspekt befinner den forhåndsfylte sprøyten seg også i en injeksjonsanordning. I et ytterligere spesifikt aspekt er injeksjonsanordningen en autoinjektor I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen et antistoff ifølge oppfinnelsen for anvendelse i en fremgangsmåte for spesifikk desimering av IgEproduserende B-celler, som omfatter å administrere en terapeutisk effektiv mengde av et antistoff mot IgE/M1' som spesifikt binder M1'-segmentet av IgE og utløser apoptose i IgE-uttrykkende B-celler som definert i kravene. I et spesifikt aspekt omfatter antistoffet HVR i den tunge og lette kjeden til antistoffet som fremkommer i en hvilken som helst av figurene 6A 6F. I et annet spesifikt aspekt senker fremgangsmåten det totale serum-ige. I nok et annet spesifikt aspekt senker fremgangsmåten både fritt og totalt serum-ige. I et ytterligere spesifikt aspekt er serum-ige allergenspesifikt. I nok et ytterligere spesifikt aspekt velges antistoffet fra gruppen som består av: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. I enda et ytterligere spesifikt aspekt er antistoffet 47H4v. I enda et ytterligere spesifikt aspekt har antistoffet ADCC-aktivitet.
.")
9 I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen et antistoff ifølge oppfinnelsen til anvendelse i en fremgangsmåte for å behandle en IgE-formidlet forstyrrelse, som omfatter å administrere en terapeutisk effektiv mengde av et antistoff mot IgE/M1' som spesifikt binder M1'-segmentet av IgE og utløser apoptose i IgE-uttrykkende B-celler som definert i kravene. I et spesifikt aspekt desimerer antistoffet spesifikt IgE-produserende B-celler. I et annet spesifikt aspekt, senker antistoffet det total serum-ige. I nok et annet spesifikt aspekt senker antistoffet både totalt og fritt serum-ige. I et ytterligere spesifikt aspekt er serum-ige allergenspesifikt. I enda et ytterligere spesifikt aspekt omfatter antistoffet HVR i den tunge og den lette kjeden til antistoffet som fremkommer i en hvilken som helst av figurene 6A 6F. I nok et ytterligere spesifikt aspekt velges antistoffet fra gruppen som består av: 26A11, 26A11 vl-16, 7A6, 7A6v1, 47H4, 47H4v1-6. I et ytterligere spesifikt aspekt er antistoffet 47H4v. I enda et ytterligere spesifikt aspekt har antistoffet ADCC-aktivitet. I enda et ytterligere spesifikt aspekt velges den IgE-formidlede forstyrrelsen fra gruppen som består av: allergisk rhinitt, astma (f.eks. allergisk astma og ikke-allergisk astma), atopisk dermatitt, allergisk gastroenteritt, overfølsomhet (f.eks. anafylaksi, elveblest, matallergier osv.), allergisk bronkopulmonal aspergillose, parasittsykdommer, interstitiell cystitt, hyper-ige-syndrom, ataksi-telangiektasi, Wiskott-Aldrich-syndrom, lymphoplasia athymica, IgE-myelom og transplantatmot-vert-reaksjon. I nok et ytterligere spesifikt aspekt er den IgE-formidlede forstyrrelsen matallergi, anafylaksi, kontaktdermatitt og allergisk purpura I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen et antistoff ifølge oppfinnelsen for anvendelse i en fremgangsmåte for å behandle en IgEformidlet forstyrrelse, som omfatter å administrere en sammensetning som omfatter en terapeutisk effektiv mengde av et antistoff mot IgE/M1' som spesifikt binder M1'-segmentet av IgE, og utløser apoptose i IgE-uttrykkende B-celler, i kombinasjon med en terapeutisk effektiv mengde av minst ett legemiddel valgt fra gruppen som består av: antistoff mot IgE, antihistamin, bronkodilator, glukokortikoid, NSAID, slimhinneavsvellende middel, hostedempende legemiddel, analgetikum, TNF-antagonist, integrinantagonist, immunundertrykkende middel, IL-4-antagonist, IL-13-antagonist, dobbel IL- 4/IL-13-antagonist, DMARD, antistoff som binder til en B-celleoverflatemarkør og BAFF-antagonist som definert i kravene. I et spesifikt aspekt desimerer antistoffet spesifikt IgE-produserende B-celler. I et annet spesifikt aspekt, senker antistoffet det total serum-ige. I nok et annet spesifikt aspekt senker

10 8 antistoffet både totalt og fritt serum-ige. I et ytterligere spesifikt aspekt er serum-ige allergenspesifikt. I enda et ytterligere spesifikt aspekt omfatter antistoffet HVR i den tunge og den lette kjeden til antistoffet som fremkommer i en hvilken som helst av figurene 6A 6F. I nok et ytterligere spesifikt aspekt velges antistoffet fra gruppen som består av: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. I enda et ytterligere spesifikt aspekt er antistoffet 47H4v. I enda et ytterligere spesifikt aspekt har antistoffet ADCC-aktivitet I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen et antistoff ifølge oppfinnelsen for anvendelse i en fremgangsmåte for å behandle en IgE-formidlet forstyrrelse som omfatter et kombinert behandlingsregime for å administrere en terapeutisk effektiv mengde av et antistoff mot IgE/M1' som spesifikt binder M1 -segmentet av IgE og utløser apoptose i IgE-uttrykkende B-celler, før, samtidig med eller etter administrering av en kjent fremgangsmåte for å behandle allergiske forstyrrelser som definert i kravene. I et spesifikt aspekt omfatter kombinasjonen å administrere et antistoff mot IgE, antihistamin, en bronkodilator, et glukokortikoid, et ikke-steroidisk antiinflammatorisk legemiddel, et immunundertrykkende middel, IL-4-antagonist, IL-13- antagonister, dobbel IL-4/IL-13-antagonist, et slimhinneavsvellende middel, et hostedempende legemiddel eller et analgetikum. I et annet spesifikt aspekt administreres antistoff mot IgE/M1'et i kombinasjon med et behandlingsregime for desensibilisering mot allergener. I et spesifikt aspekt desimerer antistoffet spesifikt IgE-produserende B-celler. I et annet spesifikt aspekt, senker antistoffet det total serum-ige. I nok et annet spesifikt aspekt senker antistoffet både totalt og fritt serum-ige. I et ytterligere spesifikt aspekt er serum-ige allergenspesifikt. I enda et ytterligere spesifikt aspekt omfatter antistoffet HVR i den tunge og den lette kjeden til antistoffet som fremkommer i en hvilken som helst av figurene 6A 6F. I nok et ytterligere spesifikt aspekt velges antistoffet fra gruppen som består av: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. I enda et ytterligere spesifikt aspekt er antistoffet 47H4v. I enda et ytterligere spesifikt aspekt har antistoffet ADCC-aktivitet. 3 I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen et antistoff ifølge oppfinnelsen for anvendelse i en fremgangsmåte for å behandle allergenindusert IgE-produksjon som omfatter å administrere en terapeutisk effektiv mengde av et antistoff mot IgE/M1' som spesifikt binder M1'-segmentet av IgE, og utløser apoptose i IgE-uttrykkende B-celler som definert i kravene. I et spesifikt aspekt

11 omfatter antistoffet HVR i den tunge og lette kjeden til antistoffet som fremkommer i en hvilken som helst av figurene 6A 6F. I et annet spesifikt aspekt desimerer fremgangsmåten spesifikt IgE-produserende B-celler. I nok et annet spesifikt aspekt senker fremgangsmåten det totale serum-ige. I et ytterligere annet spesifikt aspekt senker fremgangsmåten både fritt og totalt serum-ige. I enda et ytterligere spesifikt aspekt er serum-ige allergenspesifikt. I nok et ytterligere spesifikt aspekt velges antistoffet fra gruppen som består av: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. I enda et ytterligere spesifikt aspekt er antistoffet 47H4v. I enda et ytterligere spesifikt aspekt har antistoffet ADCC-aktivitet. I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen et antistoff ifølge oppfinnelsen for anvendelse i en fremgangsmåte for å redusere den allergenutløste IgE-produksjonen, som omfatter å administrere en terapeutisk effektiv mengde av et antistoff mot IgE/M1' som spesifikt binder M1'-segmentet av IgE og utløser apoptose i IgE-uttrykkende B-celler som definert i kravene. I et spesifikt aspekt omfatter antistoffet HVR i den tunge og lette kjeden til antistoffet som fremkommer i en hvilken som helst av figurene 6A 6F. I et annet spesifikt aspekt desimerer fremgangsmåten spesifikt IgE-produserende B-celler. I nok et annet spesifikt aspekt senker fremgangsmåten totalserum-ige. I et ytterligere annet spesifikt aspekt senker fremgangsmåten både fritt og totalt serum-ige. I enda et ytterligere spesifikt aspekt er serum-ige allergenspesifikt. I enda et ytterligere spesifikt aspekt velges antistoffet fra gruppen som består av: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. I enda et ytterligere spesifikt aspekt er antistoffet 47H4v. I enda et ytterligere spesifikt aspekt har antistoffet ADCC-aktivitet. I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen en sammensetning som kan brukes til en hvilken som helst av de tidligere beskrevne fremgangsmåtene, som definert i kravene. 30 I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen anvendelse av en sammensetning i en hvilken som helst av de tidligere beskrevne fremgangsmåtene som definert i kravene. 3 I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen et murint hybridom deponert i ATCC den 21. mars 2007 med en betegnelse valgt fra gruppen som består av: 7A6.18, IC11..20, 47G4.6.2, 47H4.12., 42H4.6.9, 42A.20.11,

12 26A11.6., 1D2.22.1, 4C1.6.14, 26B , 28E I et spesifikt aspekt tilveiebringer oppfinnelsen et antistoff utskilt av det deponerte hybridomet. I enda en ytterligere utførelsesform tilveiebringer oppfinnelsen et transgent dyr som uttrykker det humane M1'-segmentet som definert i kravene. KORT BESKRIVELSE AV TEGNINGENE Figur 1A B er en sammenstilling av valgte konstantkjederegioner av IgE fra menneske (SEQ ID NO:1), rhesusape (SEQ ID NO:2) og krabbemakak (SEQ ID NO:3). Figuren viser de omtrentlige posisjonene til de transmembrandomenene og intracellulære domenene CH2, CH3, CH4, M1' Figur 2A 2C er FACS Scatchard-plott som viser spesifisiteten til de ulike anti- M1 antistoffene. Figur 2A-1 til 2A-6 viser binding mot den korte formen av IgE (mangler M1'), mens figurene 2B-1 til 2B-6 viser binding mot den lange formen (M1'). Figur 2C-1 til 2C-6 viser binding mot IgE uttrykt av U266-cellestammen. Den skyggelagte kurven viser fluorescensintensiteten til kontroll-antistoffet, mens den ikke-skyggelagte kurven viser den relative fluorescensen til det testede antistoffet. Figur 2D F viser bindingsspesifisitetene til de murine antistoffene mot IgE/M1, 47H4, 26A11 og 7A6. Figur 2D viser at 47H4 binder IgE/M1 fra menneske, rhesusape og krabbemakak, men ikke IgE som mangler M1'. Figur 2E viser at 47H4 binder U266, mens 26A11 og 7A6 ikke gjør det. Figur 2F viser at 47H4 og 7A6 binder både rhesus- og krabbemakak-m1', mens 26A11 bare binder rhesus. Figur 2G-1 viser bindingsspesifisitetene til de humaniserte antistoffene mot IgE/M1, 47H4v, 2611v6 og 7A6v1. Figur 2G viser at alle de tre humaniserte variantene 47H4v, 26A11v6 og 7A6v1 er spesifikke for IgE-M1', men ikke for IgE som mangler M1'. Figur 2H viser at variantene 47H4v og 26A11v6 (men ikke 7A6v1) binder U266. Figur 2I viser at 47H4v og 7A6v1 binder rhesus- og krabbemakak-m1', mens 26a11v6 bare binder rhesus-m1'. 3 Figur 3A-L er FACS-plott som viser de relative bindingsaffinitetene til de forskjellige anti-m1'-antistoffene ved hjelp av seriefortynningene som er angitt i figur 3M. Figur 3N viser den relative affiniteten for hvert antistoff. Figur 3O oppsummerer affiniteten til de murine antistoffene 47H4, 26A11 og 7A6 målt ved Scatchardanalyse mot human, rhesus- og krabbemakak-m1'. Med mindre det er angitt noe annet, betyr de oppgitte tallene aritmetiske gjennomsnitt. Figur 3P oppsummerer

13 11 affiniteten til de angitte humaniserte variantene av antistoffene 47H4 og 26A11, målt ved Scatchard-analyse mot human, rhesus- og krabbemakak-m1'. Figur 4A-D viser en relativ bindings-/blokkeringsstudie av antistoffer mot M1'. Figur 4A-1 til 4A-20 er FACS-plott som viser antistoffer som blokkerte eller delvis blokkerte bindingen. Figur 4B er et todimensjonalt plott som viser den relative evnen til å blokkere binding av de andre antistoffene (delvis eller fullstendig ved å anvende et molforhold på 1:1). Figur 4C er et todimensjonalt plott som er mer fokusert på de spesifikt angitte antistoffene og der blokkeringsstudien ble gjentatt med et molforhold på :1. Figur 4D er et skjema som viser grupperingene som følger av epitopbindings-/blokkeringsstudiene Figur A C viser epitopbindingsstudier utført ved hjelp av 47H4, 7A6 og 26A11. Figur A viser M1'-segmentet inkludert tilstøtende N- og C-terminale rester (SEQ ID NO:3) og M1'-peptidene 1-1, (SEQ ID NO: 19), som anvendes til å bestemme epitopbindingen. Figur B og D viser at de murine opphavsantistoffene 47H4 binder peptid 4, 7A6 binder peptid 4 og og 26A11 binder peptidene 7 og 8. Figur C og E viser at de humaniserte variantene 47H4v binder peptid 4, mens 7A67v1 binder peptidene 4 og, og 26A11v6 binder peptidene 7 og 8 og beholder dermed epitopspesifisitetene til de murine opphavsantistoffene Figur 6A F viser de variable lette og tunge kjedesekvensene til de murine antistoffene 26A11, 7A6 og 47H4 og forskjellige humaniserte varianter av disse. Posisjonene er nummerert ifølge Kabat og de hypervariable regionene som ble podet på det variable konsensusnettverket (kappa I for lett kjede, undergruppe III for tung kjede) er plassert i firkanter. Figur 6A viser, i forhold til human kappa I lett kjede (SEQ ID NO:20), den variable lette kjeden til 26A11 (SEQ ID NO:21) og de humaniserte variantene 1, 4 (SEQ ID NO:22), variant 2, (SEQ ID NO:23), variant 3, 6 (SEQ ID NO:24), variant 13, 1 (SEQ ID NO:2) og variant 14, 16 (SEQ ID NO:26). Figur 6B viser, i forhold til human kappa I lett kjede (SEQ ID NO:20), den variable lette kjeden til 7A6 (SEQ ID NO:27) og den humaniserte variant I (SEQ ID NO:28). Figur 6C viser, i forhold til human kappa I lett kjede (SEQ ID NO:20), den variable lette kjeden til 47H4 (SEQ ID NO:29) og de humaniserte variantene 1, 3 (SEQ ID NO:30) og variantene 2, 4 6 (SEQ ID NO:31). Figur 6D viser, i forhold til den humane tunge kjeden (SEQ ID NO:32), den variable tunge kjeden til 26A11 (SEKV ID NO: 33) og de humaniserte variantene 1 3, 13, 14 (SEQ ID NO:34) og variantene 4 6, 1, 16

14 12 (SEQ ID NO:3). Figur 6E viser, i forhold til den humane tunge kjeden (SEQ ID NO:32), den variable tunge kjeden av 7A6 (SEQ ID NO:33) og den humaniserte varianten 1 (SEQ ID NO:37). Figur 6F viser, i forhold til den humane tunge kjeden III (SEQ ID NO:32), den variable tunge kjeden til 47H4 (SEQ ID NO:38), og de humaniserte variantene 1,2 (SEQ ID NO: 39), variantene 3 4 (SEQ ID NO: 40), variant (SEQ ID NO: 41) og variant 6 (SEQ ID NO:42) Figur 7A G viser den apoptotiske aktiviteten til de parenterale M1'-antistoffene i IgE-M1'-transfekterte Daudi-celler. Figur 7A er et FACS-plott som viser at IgM i det vesentlige uttrykkes på et høyere konsentrasjon enn IgE. Figur 7B viser virkningen av å fornette anti-igm [F(ab')2]-antistoffer og anvendelse av kamptotecin til å utløse apoptose. Cellene som farger positivt for annexin, men negativt for PI, er døende, mens cellene som farger positivt for både annexin og PI, er døde. Figur 7C er en grafisk fremstilling av den totalt observerte apoptosen, der den lyse linjen viser cellenes annexin-(+) og PI-(-), mens den mørke linjen viser annexin-(+) og PI-(+). Figur 7D 7G er grafiske fremstillinger av anti-m1'-utløst apoptose ved konsentrasjoner på 2,, 1, 0,1, 0,01 og 0,001 µg/ml. Figur 7D viser resultatene av anvendelsen av M1'-antistoffene av epitopgruppen A1, inkludert 7A6, 47H4, 47G4, 42A, 42H4 (sammen med kontrollene MAE-11 og GP120) uten kryssbinding. Figur 7E viser resultatene av anvendelsen av M1'-antistoffene av epitopgruppen A2, inkludert IC11, 26A11, 1D2, 4C1) og epitopgruppen B/C, inkludert 26B11 og 28E9 uten kryssbinding. Figur 7F viser epitopgruppe A1 med kryssbinding, og Figur 7G viser epitopgruppe A2 og B/C med kryssbinding Figur 8A B viser den apoptotiske aktiviteten til de humaniserte anti-m1'- variantene i Daudi-celler transfektert med IgE-M1' behandlet med forskjellige humaniserte anti-m1'-antistoffvarianter i konsentrasjoner på 2,, 1, 0,1, 0,01 og 0,001 µg/ml. Figur 8A viser at de humaniserte variantene 47H4v, 26A11v6 og 7A6v1 utløser apoptose i størrelsesorden % ved de høyere konsentrasjonskonsentrasjonene (dvs. 2 µg/ml for 47H4- og 26A11- variantene, 1, og 2 µg for 7A6-varianten). Figur 8B viser den apoptotiske aktiviteten til det samme antistoffet i de IgE-M1 -transfekterte Daudi-cellene som behandles i nærvær av det kaprine antihumane IgG F(ab )2-kryssbundne antistoffet. Alle antistoffene utløste maksimalt apoptotisk konsentrasjon på %, med en viss nedgang i apoptoseaktiviteten ved høy konsentrasjon (f.eks. 47H4 v1, v2, 26A11 v1, v14). Figur 8C viser at både naturlig og afukosylert 47H4v kunne utløse apoptose ved tilsvarende konsentrasjon.

15 13 Figur 9A B1-2 viser de murine M1-antistoffenes evne til å utløse kalsiumfluks i Daudi-IgE/M1'-celler. Figur 9A er kontrollen som viser virkningen av anti-igm og MAE11, mens Figur 9B1 B2 viser virkningen av anvendelsen av de angitte murine anti-m1'-antistoffene. Figur viser det humaniserte IgE/M1 -antistoffet 47H4 v wt og den afukosylerte varianten sin evne til å utløse ADCC. De wt- og afukosylerte variantene utløser tilsvarende maksimal cytotoksisitet, men den afukosylerte varianten («AF») var sterkere enn den naturlige formen (EC 0 AF 0,83 nm, EC 0 wt 6,6 nm) Figur 11A 11F er fullengdesekvensene til den tunge og lette kjeden av de murine IgE/M1-antistoffene. De variable regionene er vist i kursiv, mens HVR (de hypervariable regionene) er understreket. Figur 11A viser den tunge og lette kjeden til det murine antistoffet 7A6, henholdsvis (SEQ ID NO: 43 og 44). Figur 11B viser den tunge og den lette kjeden til det murine antistoffet 47H4, henholdsvis (SEQ ID NO: 4 og 46). Figur 11C viser den tunge og den lette kjeden til det murine antistoffet 26A11, henholdsvis (SEQ ID NO: 47 og 48). Figur 11D viser den tunge og den kjeden til det murine antistoffet 4C1, henholdsvis (SEQ ID NO: 49 og 0). Figur 11E viser den tunge og den lette kjeden til det murine antistoffet 28E9, henholdsvis (SEQ ID NO: 1 og 2). Figur 11F viser den tunge og den lette kjeden til det murine antistoffet 1C11, henholdsvis (SEQ ID NO: 3 og 4) Figur 12A I demonstrerer de murine anti-ige/m1-antistoffenes evne til å hemme produksjonen av serum-ige og IgE-produserende plasmaceller i en atopisk hu-scid-modell. Figur 12A er en grafisk fremstilling av forsøksutformingen. Figur 12B C viser at behandling med de murine antistoffene mot IgE/M1' reduserte IgE-konsentrasjonen med 6 til 84 %. Figur 12D E viser at desimeringen av IgE-produserende celler in vivo var %. Figur 12F G viser at konsentrasjonen av andre immunoglobuliner (f.eks. IgG1-4, IgA, IgM) var relativt upåvirket. Figur 12H I viser at det ikke ble observert noen reduksjon i det totale antallet plasmaceller i milten. 3 Figur 13A H viser virkningen av den humaniserte varianten 47H4v på immunoglobulinkonsentrasjonen i den atopiske hu-scid-modellen. Figur 13A er en grafisk fremstilling av forsøksutformingen. Figur 13B D viser at serum- IgE ble senket med 79 %, og at konsentrasjonen av de IgE= produserende
 var sterkere enn den naturlige formen (EC 0 AF 0,83 nm, EC 0 wt 6,6 nm).")
16 14 plasmacellene ble senket med 7 %. Figur 13E F viser ingen senket konsentrasjon av andre serumimmunoglobuliner. Figur 13G H viser ingen senking av den totale plamacellekonsentrasjonen og demonstrerer dermed at 47H4 spesifikt desimerer de IgE-produserende plasmacellene, som er en svært liten andel av de totale plasmacellene Figur 14A D illustrerer produksjon av hum1'-knock-in-mus. Figur 14A viser plasseringen av M1 -eksonet på IgE-lokusen til musen. Figur 14B viser et skjema av rekombinasjonen i IgE-lokusen til musen som fører til at målallelet dannes. Figur 14C viser PCR-genotyping av et 668 bp bånd hos wt-musene og et 47 bp bånd hos M1'-«knock-in»-musene. Figur 14D viser et southern blot der et 7,4 kb HindIII-fragment hos wt-musene blir et 3 kb fragment hos hu- M1'-knockins-musene, og et 14,1 kb BamHI-fragment blir et 18,1-fragment i hu-m1'-knockinallelet. Figuren viser både naturlige og heterozygote mus. Figur 1A-I viser IgE/M1 -antistoffenes evne til å hindre produksjon av IgE i en primær immunrespons. Figur 1A er et skjema som viser tidslinjen til forsøksutformingen, inkludert administrering av TNP/OVA og antistoff mot IgE/M1'. Figur 1B er en graf av antigenspesifikke IgE-konsentrasjoner over tid, og viser at mens antigenspesifikke IgE-konsentrasjoner hos kontrolldyrene (dvs. gp120) nådde maksimale konsentrasjoner mellom dag 8 og 14, hindret anti-ige/m1' enhver økning, og de målte antigenspesifikke IgE-konsentrasjonene var ikke signifikant forskjellig fra de ikke-immuniserte musene. Figur 1C D viser at IgE/M1 -antistoffbehandlingen hindret det antigenspesifikke serumet IgE i å øke på dag 8 og 14, og var ikke statistisk forskjellig fra de uimmuniserte musene (Figur 1E). Figur 1F I viser at konsentrasjonen av antigenspesifikt IgG1 ikke ble signifikant påvirket av anti-ige/m1' i løpet av de 28 forsøksdagene (med unntak for en beskjeden forskjell på dag 14) Figur 16A-K viser IgE/M1 -antistoffenes evne til å forhindre produksjon av antigenspesifikk IgE i en minnerespons eller sekundær immunrespons. Figur 16A er et skjema som viser tidslinjene for sekundær økning av TNP-OVA og administrering av IgE/M1 -antistoffene, som ble gitt først på dag 28. Figur 16B er en graf som viser de antigenspesifikke IgE-konsentrasjonene over tid, og viser at den sekundære IgE-responsen på TNP-OVA-økningen på dag 28 er hurtigere, med en topp etter 4 dager i stedet for 8 9 dager i den primære responsen. Figur 16C- D viser at de antigenspesifikke IgE-konsentrasjonene i de anti-ige/m1'-

17 1 1 behandlede dyrene ble signifikant redusert sammenlignet med isotypekontrollen, 9 6 % på dag 32 og % på dag 3. Figur 16E viser at på dag 42 (12 dager etter første administrering med anti-ige), ble de antigenspesifikke IgEkonsentrasjonene redusert til en konsentrasjon som ikke var signifikant forskjellig fra de naive kontrollmusene. Figur 16F H viser at mellom dag 28 og 49, reduserte administreringen av anti-ige/m1' serumkonsentrasjonen av IgE med %, og den gjennomsnittlige daglige konsentrasjonen av antigenspesifikk IgE ble også redusert med %. Figur 16J K viser at konsentrasjonen av antigenspesifikk IgG1 ikke ble signifikant påvirket av anti-ige/m1'. Figur 17A D illustrerer IgE/M1 -antistoffenes evne til forebyggende reduksjon av produksjonen av IgE som respons på Nippostrongylus brasiliensis («NB»)- infeksjonen. Figur 17A er et skjema som viser forsøksutformingen. Dyrene ble behandlet tre ganger per uke med start på dag 0 og ut dag 21. Figur 17B viser IgE-konsentrasjonen over tid som respons på NB-infeksjon med IgE/M1 - og kontrollantistoffer. Figur 17C D viser at på dag 1 hadde anti-ige/m1'- behandlede dyr en senket serumkonsentrasjon av IgE som ikke var statistisk signifikant forskjellig fra friske mus Figur 18A I illustrerer IgE/M1 -antistoffenes evne til å gi terapeutisk behandling av IgE-toppresponsen på Nippostrongylus brasiliensis-(«nb»)- infeksjonen, Figur 18A er et skjema som viser forsøksutformingen. Dyrene ble behandlet tre ganger per uke mellom dagene 11 og 21. Figur 18B viser IgEkonsentrasjonen over tid som respons på NB-infeksjonen med anti-ige/m1' og kontrollantistoffer. Figur 18C D viser at anti-ige/m1' senket serumkonsentrasjonen av IgE med % i løpet av fire dagers behandling. Figur 18E viser at på dag 21 var IgE-konsentrasjonen i de anti-ige/m1'- behandlede senket med %, og de oppnådde konsentrasjonene var ikke statistisk signifikant forskjellige fra den ikke-infiserte kontrollgruppen. Figur 18F G viser at de IgE-produserende plasmacellene (kvantifisert ved Elispot) i lymfeknutene og milten var desimert med henholdsvis % og 7 66 %. Figur 18H I viser at de totale plasmacellene (CD138+) i både lymfeknutene og milten økte i alle behandlingsgruppene sammenlignet med de ikke-infiserte musene, og at behandling med anti-ige/m1' ikke signifikant endret det totale antallet plasmaceller i noen av organene. Disse resultatene demonstrerer IgE/M1-antistoffenes evne til å senke serumkonsentrasjonen av IgE ved å desimere de IgE-produserende cellene in vivo.

18 16 Figur 19A G illustrerer IgE/M1 -antistoffenes evne til terapeutisk behandling av IgE-responsen som opptrer sent i en infeksjonssyklus mot Nippostrongylus brasiliensis («NB»)-infeksjonen. Figur 19A er et skjema som viser forsøksutformingen der dyrene ble behandlet tre ganger per uke med start på dag 40. Figur 19B viser at topproduksjonen av IgE kom rundt dag 1, og at alle IgE/M1 -antistoffene senket serum-ige. Figur 19C D viser at IgE/M1 - antistoffet gir signifikant senkning av både den absolutte og den normaliserte IgE-konsentrasjonen sammenliknet med behandlingsstarten. Figur 19E-G viser at anti-ige/m1'-behandlingen signifikant senket serumkonsentrasjonen av IgE, sammenlignet med anti-gp120 migg1-isotypekontrollen mellom dag 48 og. DETALJERT BESKRIVELSE AV DEN FORETRUKNE UTFØRELSESFORMEN 1 Generelle metoder 20 2 Utøvelsen av den foreliggende oppfinnelsen vil anvende, med mindre annet er indikert, konvensjonelle metoder i molekylærbiologi (inkludert rekombinante metoder), mikrobiologi, cellebiologi, biokjemi og immunologi, som er innenfor teknikkens stand. Slike metoder er forklart fullstendig i litteraturen, som f.eks. Molecular Cloning: A Laboratory Manual, andre utgave (Sambrook et al., 1989); Oligonucleotide Synthesis (M.J. Gait, ed., 1984); Animals Cell Culture (R.I. Freshney, utg., 1987); Methods in Enzymology (Academic Press, Inc.); Current Protocols in Molecular Biology (F.M. Ausubel et al., utg og periodiske oppdateringer); PCR: The Polymerase Chain Reaction, (Mullis et al., utg., 1994); A Practical Guide to Molecular Cloning (Perbal Bernard V., 1988); Phage Display: A Laboratory Manual (Barbas et al., 2001) Lymfocyttutvikling og -aktivering De to hovedtypene av lymfocytter hos mennesker er T (tymus-avledet) og B (beinmargavledet). Disse cellene avledes fra de hematopoetiske stamcellene i beinmargen og fosterleveren som har spesialisert seg til den lymfoide utviklingsveien. Avkommet til disse stamcellene følger divergerende veier når de modnes til enten B- eller T-lymfocytter. Den humane B-lymfocyttutviklingen foregår i sin helhet i beinmargen. T-celler utvikles derimot fra de umodne

19 17 forløperne som forlater beinmargen og går gjennom blodstrømmen til brisselen, der de mangfoldiggjør seg og differensierer til modne T-lymfocytter. 1 De modne lymfocyttene som dukker opp i brisselen eller beinmargen er i en passiv tilstand eller «hvile»-tilstand, dvs. at de er mitotisk inaktive. Når de spres ut i blodstrømmen går disse «naive» lymfocyttene inn i forskjellige sekundære eller perifere lymfoide organer, som f.eks. milten, lymfeknutene eller mandlene. De fleste naive lymfocyttene har i seg selv kort levetid og dør noen få dager etter de har forlatt beinmargen eller brisselen. Men hvis en slik celle mottar signaler som tyder på nærvær av et antigen, kan de aktiveres og gjennomgå flere runder med celledeling. Noen av de resulterende avkomcellene går deretter tilbake til hviletilstanden og blir til minnelymfocytter B- og T-celler som kan sies å være primet for det neste møtet med allergenet som stimulerte dem. Det andre avkommet av aktiverte jomfrulymfocytter er effektorceller, som overlever i bare noen få dager, men utfører spesifikke defensive aktiviteter. 20 Lymfocyttaktivering handler om en ordnet rekke hendelser som en hvilende lymfocytt passerer gjennom når den stimuleres til å dele seg og produsere avkom, og noen av dem blir effektorceller. En fullstendig respons inkluderer både utløsningen av celleproliferasjonen (mitogenese) og uttrykkingen av immunologiske funksjoner. Lymfocyttene aktiveres når spesifikke ligander binder til reseptorene på overflatene deres. Ligandene er forskjellige for T-celler og B- celler, men de resulterende intracellulære fysiologiske mekanismene er like Fremmede antigener kan selv utløse lymfocyttaktivering, spesielt store polymerantigener som kryssbundne overflateimmunoglobuliner på B-celler eller andre glykoproteiner på T-celler. De fleste antigener er imidlertid ikke polymere, og selv ved direkte binding til B-cellen i et stort antall fører det ikke til aktivering. B-cellene aktiveres av disse mer vanlige antigenene når de stimuleres sammen med nesten aktiverte hjelpe-t-lymfocytter. Slik stimulering kan stamme fra lymfokiner som skilles ut av T-cellen, men den overføres mest effektivt ved direkte kontakt mellom B-cellen og T-cellens overflateproteiner som interagerer med visse overflatereseptorer på B-cellen og danner et sekundært signal. 3 B-celler

20 18 Den definerende egenskapen til B-celler er evnen til å syntetisere immunoglobuliner. Immunoglobuliner (Ig) er en ekstremt mangfoldig familie av proteiner som består av beslektede typer polypeptider kalt tunge kjeder og lette kjeder. Hvert Ig binder spesifikt sitt eget spesifikke antigen med høy affinitet. Modne B-celler kan uttrykke immunoglobulin i to forskjellige former, som hver tjener unike funksjoner. I hvilende B-lymfocytter (naive eller minne) uttrykkes immunoglobuliner bare på celleoverflaten, der de opptrer hovedsakelig som membranbundet reseptor for spesifikke antigener. Derimot skiller B- celleeffektorceller (plasmaceller) ut immunoglobulin i omgivelsene. Slikt utskilt immunoglobulin beholder evnen til å gjenkjenne og binde (side ved side med den membranbundne formen på hvilende B-celler), og kalles vanligvis antistoffer Når en aktivert B-lymfocytt deler seg, blir noe av avkommet minne-b-celler, mens de øvrige differensierer til plasmaceller. Ettersom plasmaceller har relativt kort levetid, dør populasjonen snart ut med mindre det produseres nye plasmaceller, og det skilles ikke ut immunoglobuliner lenger. Derfor fører aktivering av B-celler vanligvis til en forbigående prolifereringsbølge, som så følges av et utbrudd av antistoffutskilling som øker og deretter avtar i løpet av flere dager eller noen få uker. B-celler er den primære celletypen som er involvert i humoral immunitet, eller den beskyttende virkningen som formidles gjennom vevsvæsker. Ettersom anti-ige/m1'-antistoffene ifølge oppfinnelsen faktisk desimerer B-celler, inkludert minne-b-celler, kan de anvendes til å «tilbakestille» hukommelsen. Virkningen av dette kan dermed være at B-cellekomponenten som driver den allergiske responsen hos individer kan dempes, om ikke elimineres. T-celler 30 T-lymfocytter uttrykker ikke immunoglobuliner, men registrere i stedet nærværet av fremmede stoffer ved hjelp av overflateproteiner som kalles T-cellereseptorer. Disse reseptorene gjenkjenner antigener enten ved direkte kontakt eller ved å påvirke aktiviteten til andre immunceller. Sammen med makrofagene er T-cellene den primære celletypen som er involvert i den celleformidlede immuniteten. 3 I motsetning til B-cellene kan T-cellene registrere fremmede stoffer i spesifikke sammenhenger. Nærmere bestemt vil T-lymfocyttene gjenkjenne et fremmed protein bare hvis det først spaltes til små peptider, som deretter vises på overflaten av en andre vertscelle, kalt en antigenpresenterende celle (APC). En
 uttrykkes immunoglobuliner bare på celleoverflaten, der de opptrer hovedsakelig som membranbundet reseptor for spesifikke antigener.")
21 19 hvilken som helst type vertscelle kan presentere antigener under enkelte forhold, men visse typer er mer spesifikt tilpasset for dette formålet, og er spesielt viktig for å kontrollere T-celleaktiviteten, blant annet makrofager og andre B-celler. Antigenpresentasjon avhenger delvis av spesifikke proteiner, kalt store histokompatibilitetskompleksproteiner (MHC), på overflaten av de presenterende cellene. For å stimulere den celleformidlede immuniteten må dermed fremmede peptider presenteres for T-celler i kombinasjon med MHCpeptider, og denne kombinasjonen må gjenkjennes av en T-cellereseptor Det er to signifikante T-celleundergrupper: cytotoksiske T-lymfocytter (T c -celler eller CTL-er) og hjelper-t-celler (T H )-celler, som grovt sett kan identifiseres på grunnlag av celleoverflateuttrykking av markøren CD8 og CD4. Tc-celler er viktige i virusforsvar, og kan drepe virus direkte ved å gjenkjenne visse celleoverflateuttrykte viruspeptider. T H -celler fremmer proliferering, modning og immunologisk funksjon av andre celletyper, f.eks. lymfokinutskilling for å kontrollere aktiviteten til B-celler, makrofager og cytotoksiske T-celler. Både naive og minne-t-lymfocytter forblir vanligvis i hviletilstanden, og i denne tilstanden viser de ikke signifikant hjelpeaktivitet eller cytotoksisk aktivitet. Når de aktiveres, gjennomgår disse cellene flere runder med mitotisk deling for å produsere datterceller. Noen av disse dattercellene går tilbake til hviletilstanden som minneceller, mens andre blir effektorceller som aktivt uttrykker hjelpere av den cytotoksiske aktiviteten. Disse dattercellene ligner opphavet sitt: CD4+-celler kan bare produsere CD4+-avkom, mens CD8+-celler bare gir CD8+-avkom. Effektor- T-celler uttrykker celleoverflatemarkører som ikke uttrykkes på hvilende T-celler, som f.eks. CD2, CD28, CD29, CD40L, transferrinreseptorer og MHC-proteiner av klasse II. Når den aktiverende stimulansen er trukket tilbake, avtar den cytotoksiske eller hjelperaktiviteten gradvis gjennom flere dager etter hvert som effektorcellene enten dør eller går tilbake til hviletilstanden På samme måte som B-celleaktiveringen, krever også T-lymfocyttrespons på de fleste antigener to typer samtidig stimulans. Den første er antigenet, som kan gjenkjennes og bindes av T-cellereseptorer hvis de vises på riktig måte av MHCproteiner på en antigenpresenterende celle. Dette antigen-mhc-komplekset sender et signal til cellens indre, men det er vanligvis ikke tilstrekkelig til å aktivere T- cellene. Full aktivering, som f.eks. skjer med hjelpe-t-celler, krever samtidig stimulering med andre spesifikke ligander som kalles kostimulatorer og uttrykkes på
22 20 overflaten av den antigenpresenterende cellen. Aktivering av en cytotoksisk T-celle krever derimot generelt IL-2, et cytokin som skilles ut av aktiverte hjelper-t-celler. 1 Immunresponsen De tre primære funksjonelle egenskapene til pattedyrimmunsystemet som skiller det fra den andre kroppens forsvar, er: (1) spesifisitet - evnen til å gjenkjenne og respondere eller ikke å respondere individuelt blant et stort antall målmolekyler, (2) diskriminering - evnen til å skille selv fra ikke-selv, å kunne sameksistere fredelig med alle de utallige proteinene og annet organisk materiale, men likevel reagere kraftig på fremmed materiale som føres inn i kroppen, og (3) minne - evnen til å formes av erfaring slik at senere møter med et bestemt fremmed patogen vil provosere en raskere og mer kraftig respons enn ved det første møtet. Siden IgE-antagonistene ifølge oppfinnelsen utløser apoptose i IgE-bærende B- celler, forventes de å svekke eller til og med slette immunminnet om bestemte antigener. Dette ventes å være en spesiell fordel når kraftig immunrespons mot vanlige allergener er patologisk, som i tilfellet er med atopiske forstyrrelser Naive lymfocytter frigjøres kontinuerlig fra de primære lymfoidorganene til periferien, der de alle bærer overflatereseptorer som gjør det mulig å binde antigener. Antigenbinding i B-celler formidles gjennom overflatebundne immunoglobuliner, men i T-cellene formidles den de av T-cellereseptorer. Hvis de naive lymfocyttene ikke aktiveres, dør de i løpet av få dager når de har kommet ut i periferien. De som aktiveres, overlever og sprer seg, og lager datterceller som deretter kan gjennomgå ytterligere aktiverings- og spredningssykluser. Hastigheten og intensiteten til responsen på et gitt antigen bestemmes i stor grad av klonal utvelgelse: jo større populasjonen av datterceller eller kloner som er spesifikke for et bestemt antigen er, jo større er antallet celler som kan gjenkjenne og delta i immunresponsen. Hver immunrespons er en kompleks og intrikat regulert sekvens av hendelser som involverer flere celletyper. Den utløses når et immunogen trer inn i kroppen, og møter en spesialisert klasse av celler som kalles antigenpresenterende celler (APC). Disse APC-ene fanger en liten mengde av immunogenet og presenterer det i en form som kan gjenkjennes av antigenspesifikke hjelper-t-lymfocytter. Da aktiveres hjelper-t-cellene og fremmer i sin tur aktivering av andre klasser av lymfocytter, som f.eks. B-celler eller cytotoksiske T-celler. Deretter sprer de aktiverte lymfocyttene seg og utfører de spesifikke effektorfunksjoner sine. Ved
23 21 hvert trinn i denne prosessen kommuniserer lymfocyttene og APC-ene med hverandre ved direkte kontakt eller ved å skille ut regulerende cytokiner. Eksogene antigener som fanges av en APC gjennomgår en rekke endringer som kalles antigenprosessering. En slik behandling, særlig av proteinholdige immunogener involverer denaturering og delvis proteolytisk digerering slik at immunogenet spaltes til korte peptider. Et begrenset antall av de resulterende peptidene bindes deretter ikke-kovalent til MHC-proteiner av klasse II, og transporteres til APC-overflaten, en prosess som er kjent som antigenpresentering. En CD4+-hjelper-T-lymfocytt som kommer i direkte kontakt med en APC kan aktiveres, men den vil bare gjøre det hvis den uttrykte et T-cellereseptorprotein som kan gjenkjenne og binde det bestemte peptid-mhc-komplekset som presenteres av APC-en Hjelper-T (T H )-celler er hovedorganisatorene av immunresponsen siden de er nødvendige for aktivering av de to andre lymfatiske effektorcellene: cytotoksiske T (Tc)-celler og antistoffutskillende plasmaceller. TH-aktiveringen skjer tidlig i en immunrespons, og krever minst to signaler. Ett signal tilveiebringes ved å binde T-celleantigenreseptoren til det antigeniske peptid- MHC-komplekset på APC-overflaten og overføres gjennom CD3- proteinkomplekset, mens det andre, kostimulerende signalet gjennom APC-en antas å komme fra binding av et separat signaloverførende protein på T- celleoverflaten med en spesifikk ligand på APC-en. En slik kjent interaksjon er T-celleproteinet CD28 og familien av APC-overflateproteiner, kjent som B7. Andre overflateproteinpar kan også formidle kostimulering Sammen får de to signalene hjelper-t-cellen til å begynne å skille ut et cytokin, kjent som interleukin-2 (IL-2), og også til å begynne å uttrykke spesifikke IL-2- reseptorer med høy affinitet på overflaten sin. IL-2 er en svært sterk mitogen faktor for T-lymfocytter, og er avgjørende for prolifereringsresponsen til aktiverte T-celler. IL-2 virker på cellen som den skilles ut fra et fenomen kjent som en autokrin effekt. Det har også vist seg at selv om en T-celle har mottatt begge signalene, vil den ikke mangfoldiggjøre seg hvis dens egne overflatereseptorer for IL-2 blokkeres. IL-2 kan også virke på celler i umiddelbar nærhet, i en såkalt parakrin effekt. Denne effekten er spesielt viktig for å aktivere Tc-celler, som generelt ikke produserer nok IL-2 til å stimulere sin egen mangfoldiggjøring. I
24 22 tillegg til IL-2, skiller aktiverte T H -celler ut andre cytokiner og fremmer veksten, differensieringen og funksjonene til B-celler, makrofager og andre celletyper. Kontakten mellom en hvilken som helst APC og en antigenspesifikk T H -celle har også virkning på APC-en - én av de viktigste av disse er frigjøringen IL-1. Dette cytokinet antas å fungere autokrint ved å øke overflateuttrykkingen av MHC-proteiner av klasse II og av forskjellige adhesjonsmolekyler, og dermed styrke bindingen av TH-cellen og øke antigenpresentasjonen. Samtidig virker IL-1 parakrint på TH-cellen der den fremmer IL-2-utskilling og undertrykking av IL-2-reseptoren Under aktiveringen av T H -celler på den måten som er beskrevet tidligere, kan noen B-celler også ha angrepet immunogenet med antigenreseptorene sine, som er membranbundne former av antistoffene som de vil skille ut senere. I motsetning til T-celler, gjenkjenner B-celler et immunogen i sin frie, ubehandlede form. Spesifikk antigenbinding gir en type signal som kan føre til B-celleaktivering. En annen type fås fra aktiverte TH-celler, som uttrykker proteiner som bidrar til å aktivere B-cellen ved å binde seg til ikke-immunoglobulinreseptorer på overflaten av den. Disse TH-avledede signalene, som opptrer på en hvilken som helst B-celle uavhengig av antigenspesifisiteten dens, er kjent som hjelperfaktorer. Disse hjelperfaktorene inkluderer IL-2, IL-4 og IL-6. Hjelpen blir imidlertid mer effektiv ved kontakt celle til celle, noe som gjør det mulig for proteiner på T- celleoverflaten å kontakte proteinene på B-cellen direkte. Den mest effektive formen for kontaktformidlet hjelp skjer når et protein kalt CD40-liganden (CD40L), som bare uttrykkes på TH-cellene når de aktiveres, binder til et protein kalt CD40 på B-celler. I en prosess kjent som tilskueraktivering kan kontakt med en aktivert B-celle til og med være tilstrekkelig til å aktivere hvilende B-celler, selv om overflateimmunoglobulinene dens ikke har vært i kontakt med antigen Tc-lymfocytter virker ved å utrydde celler som uttrykker fremmede antigener på overflaten, som f.eks. virusinfiserte vertsceller. De fleste Tc-celler uttrykker CD8 i stedet for CD4, og gjenkjenner dermed antigener forbundet med MHC-proteiner av klasse I i stedet for MHC-proteiner av klasse II. Når en somatisk celle infiseres av et virus, kan enkelte immunogene virusproteiner ble behandlet i cellen, og de resulterende peptidene kan så dukke opp som overflatekomplekser med MHCmolekyler av klasse I. Disse peptid-mhc-kompleksene kan deretter gjenkjennes av T-cellereseptoren til en antigenspesifikk klon, som gir ett av to signaler som er
25 23 nødvendige for Tc-celleaktivering. Dette første signalet utløser alene IL-2- reseptorer med høy affinitet på Tc-cellen. Det andre signalet leveres av IL-2 som skilles ut fra en nærliggende aktivert TH-lymfocytt. Når begge signalene er mottatt, får den aktiverte Tc-cellen cytotoksisk aktivitet, som gjør at den kan drepe cellen den er bundet til, og hvilke som helst andre celler som bærer de samme MHC-peptidkompleksene av klasse I. I noen tilfeller skjer drepingen fordi Tc-en frigir spesifikke toksiner på målcellen; i andre tilfeller får Tc-en målcellen til å begå selvmord ved apoptose. Den aktiverte Tc-cellen mangfoldiggjør seg også, og gir opphav til flere Tc-celler med den samme antigenspesifisiteten. IgE/Mastcelle/Formidlingsreaksjonsvei IgE-antistoffer er festet til overflaten av mastceller og basofiler i Fc-delen av molekylet til en celleoverflatereseptor med høy affinitet, omtalt som FcεRI. Den allergiske reaksjonen starter når det flerverdige allergenmolekylet bindes til antistoffer som opptar disse reseptorene. Resultatet er en brodannelse i FcεRI, som i sin tur signalerer intracellulært og fører til frigjøring og aktivering av formidlere av inflammasjon: histamin, leukotriener, kjemotaktiske faktorer, blodplateaktiverende faktor og proteinaser. Disse aktiverte formidlerne virker lokalt og fører til økt vaskulær permeabilitet, vasodilatasjon, sammentrekking av den glatte muskulaturen og sekretutskilling fra slimkjertler. Slike aktiviteter betegnes klinisk som den umiddelbare eller tidlige fasen, og opptrer i løpet av de første 1 30 minuttene etter allergeneksponeringen. I løpet av de neste 12 timene oppstår det progressiv vevsinfiltrasjon av inflammasjonsceller, fra nøytrofiler til eosinofiler og videre til mononukleære celler som respons på andre kjemiske formidlere som vi ikke forstår fullt ut. Denne tidsperioden på 6 12 timer etter allergeneksponeringer kalles den sene fasen og er karakterisert ved kliniske manifestasjoner av celleinflammasjon. Gitt at senfasereaksjoner, særlig i lungene, kan opptre uten tidligfasereaksjoner, er det fremdeles ikke fullt forstått om den sene fasereaksjonen nødvendigvis er formidlet av IgE. 3 Denne mekanismen er primært ansvarlig for anafylaksi, elveblest og atopiske sykdommer, som f.eks. allergisk rhinitt, allergisk astma, atopisk dermatitt og allergisk gastroenteritt. Effektor-T-lymfocytten/lymfokinreaksjonsveien
26 24 Visse allergiske sykdommer formidles ved allergenreaksjon med effektor-tlymfocytten sensibilisert for det spesifikke allergenet etter en tidligere eksponering. Når den møter på allergenet, aktiveres CD4+-T-celler til å danne lymfokiner, noe som gjennom perioden på flere dager fører til opphoping av mononukleært celleinfiltrat. I. Definisjoner 1 20 Et «allergen» eller «immunogen» er et hvilket som helst molekyl som kan utløse en immunrespons. Slik den anvendes i dette dokumentet dekker betegnelsen enten det antigene molekylet i seg selv, eller kilden for det, f.eks. pollenkorn, dyreflass, insektgift eller matvarer. På den annen side har vi betegnelsen antigen, som står for et molekyl som kan gjenkjennes spesifikt av et immunoglobulin eller en T-cellereseptor. Et hvilken som helst fremmed stoff med evne til å utløse en immunrespons er et potensielt allergen. Mange forskjellige kjemikalier av både naturlig og syntetisk opphav er kjent for å være allergifremkallende. Komplekse naturlige organiske kjemikalier, spesielt proteiner, vil sannsynlig forårsake antistofformidlet allergi, mens enkle organiske forbindelser, uorganiske kjemikalier og metaller heller forårsaker T-celleformidlet allergi. I noen tilfeller kan det samme allergenet være ansvarlig for mer enn én allergitype. Eksponering for allergenet kan skje innånding, injeksjon eller hudkontakt. 2 Betegnelsen «antistoff» inkluderer monoklonale antistoffer (inkludert fullengdeantistoffer som har en immunoglobulin-fc-region), antistoffsammensetninger med polyepitopisk spesifisitet, flerspesifikke antistoffer (f.eks. bispesifikke antistoffer, dialegemer og enkeltkjedemolekyler, så vel som antistoffragmenter (f.eks. Fab, F(ab') 2 og Fv). Betegnelsene «immunoglobulin» (Ig) og «antistoff» anvendes om hverandre i dette dokumentet Den grunnleggende 4-kjedete antistoffenheten er et heterotetramert glykoprotein sammensatt av to identiske lette (L) kjeder og to identiske tunge (H) kjeder. Et IgM-antistoff består av av de grunnleggende heterotetramerenhetene sammen med et ekstra polypeptid kalt en J-kjede, og inneholder antigenbindingsseter, mens IgA-antistoffer omfatter fra 2 av de grunnleggende 4-kjedeenhetene som kan polymerisere til flerverdige konstruksjoner i kombinasjon med J-kjeden. Når det gjelder IgG-ene er 4- kjedeenheten generelt omtrent 000 dalton. Hver L-kjede kobles til en H-
27 kjede med en kovalent disulfidbinding, mens de to H-kjedene kobles til hverandre med én eller flere disulfidbindinger, avhengig av H-kjedeisotypen. Hver H- og L-kjede har også regelmessig fordelte disulfidbroer mellom kjedene. Ved N-enden har hver H-kjede et variabelt domene (V H ), fulgt av tre konstante domener (C H ) for hver av α- og γ-kjedene og fire C H -domener for µ- og ε- isotypene. Ved N-enden har hver L-kjede et variabelt domene (V L ), fulgt av et konstant domene i den andre enden. V L -et er sammenstilt med V H -en og C L -en er sammenstilt med det første konstante domenet til den tunge kjeden (C H 1). Bestemte aminosyrerester antas å danne en grenseflate mellom de variable domenene på den lette og den tunge kjeden. Sammenkoblingen av et VH og VL danner sammen et enkelt antigenbindingssete. Strukturen og egenskapene til de forskjellige antistoffklassene behandles f.eks. i Basic and Clinical Immunology, 8. utgave, Daniel P. Sties, Abba I. Terr and Tristram G. Parsolw (red.), Appleton & Lange, Norwalk, CT, 1994, side 71 og kapittel 6. L-kjeden fra en hvilken som helst virveldyrart kan tilordnes én av to klart forskjellige typer, kalt kappa og lambda, ut fra aminosyresekvensene til de konstante domenene deres. På grunnlag av aminosyresekvensen til det konstante domenet på de tunge kjedene (CH) deres, kan immunoglobuliner tilordnes forskjellige klasser eller isotyper. Det er fem immunoglobulinklasser: IgA, IgD, IgE, IgG og IgM, som har tunge kjeder betegnet henholdsvis α, δ, ε, γ og µ. γ- og α-klassene er delt ytterligere inn i underklasser på grunnlag av relativt små forskjeller i CH-sekvens og funksjon, for eksempel uttrykker mennesker de følgende underklassene: IgG1, IgG2, IgG3, IgG4, IgA1 og IgA2. Et «isolert» antistoff er et antistoff som er identifisert, separert og/eller utvunnet fra en bestanddel av produksjonsmiljøet sitt (f.eks. naturlig eller rekombinant). Det isolerte polypeptidet er fortrinnsvis uten forbindelse med noen av de andre bestanddelene av produksjonsmiljøet sitt. Forurensende bestanddeler av produksjonsmiljøet, for eksempel slike som skyldes rekombinante transfekterte celler, er materialer som vanligvis ville forstyrre forskning, diagnostikk eller terapeutisk anvendelse av antistoffet, og kan inkludere enzymer, hormoner og andre proteinholdige eller ikke-proteinholdige oppløste stoffer. I foretrukne utførelsesformer renses polypeptidet: (1) til mer enn 9 vekt-% av antistoffet f.eks. ifølge Lowry-metoden, og i noen utførelsesformer til mer enn 99 vekt-%; (2) til en grad som er tilstrekkelig til å oppnå minst 1 aminosyrerester fra N- enden eller en indre aminosyresekvens ved å anvende en roterende kopp-
28 26 sekvensator, eller (3) til homogenitet ved SDS-PAGE under ikke-reduserende eller reduserende forhold ved hjelp av Coomassie blå eller fortrinnsvis sølvfarging. Isolert antistoff inkluderer antistoffet in situ i rekombinante celler, ettersom minst én bestanddel fra antistoffets naturlige miljø ikke vil være til stede. Vanligvis vil imidlertid et isolert polypeptid eller antistoff fremstilles ved minst et rensetrinn. Den «variable regionen» og det «variable domenet» til et antistoff står for aminoendedomenene til den tunge eller lette kjeden til antistoffet. De variable domenene til den tunge kjeden og den lette kjeden kan omtales som henholdsvis «VH» og «VL». Disse domenene er generelt de mest variable delene av antistoffet (i forhold til andre antistoffer av samme klasse) og inneholder antigenbindingssetene Betegnelsen «variabel» betyr at visse segmenter av de variable domenene i stor grad varierer i sekvens fra antistoff til antistoff. V-domenet formidler antigenbinding og definerer spesifisiteten til et bestemt antistoff for antigenet det er spesifikt for. Variasjonen er imidlertid ikke jevnt fordelt over hele utstrekningen til de variable domenene. I stedet er det konsentrert til tre segmenter som kalles hypervariable regioner (HVR-er) i de variable domenene både på den lette og den tunge kjeden. De mer bevarte delene av de variable domenene kalles rammeregionene (FR). Hvert av de variable domenene til de naturlige tunge og lette kjedene omfatter fire FR-regioner, som i høy grad inntar en betaflakkonfigurasjon, forbundet med tre HVR, som danner løkker som forbinder og i noen tilfeller utgjør en del av betaflakstrukturen. HVR-ene i hver kjede holdes nært sammen av FR-regionene, og med HVR-ene fra den andre kjeden, bidrar de til å danne antigenbindingssetet for antistoffer (se Kabat et al., Sequences of Immunological Interest, femte utgave, National Institute of Health, Bethesda, MD (1991)). De konstante domenene er ikke involvert direkte i bindingen av antistoff til et antigen, men utviser forskjellige effektorfunksjoner, som f.eks. at antistoffet deltar i den antistoffavhengige celletoksisiteten. Uttrykket «monoklonalt antistoff» slik det anvendes i dette dokumentet står for et antistoff fra en populasjon av stort sett homogene antistoffer, dvs. at de individuelle antistoffene som omfatter populasjonen er identiske med unntak av mulige naturlige mutasjoner og/eller senere translasjonsmodifikasjoner (f.eks. isomerisering, amidering) som kan være til stede i mindre mengder. Monoklonale antistoffer er svært spesifikke, og rettes mot et enkelt antigensete. I motsetning til polyklonale antistoffpreparater som typisk
29 inkluderer forskjellige antistoffer rettet mot forskjellige determinanter (epitoper), rettes hvert monoklonale antistoff mot en enkelt determinant på antigenet. I tillegg til spesifisiteten sin er de monoklonale antistoffene fordelaktige ved at de syntetiseres av hybridomkulturen, uforurenset av andre immunoglobuliner. Adjektivet «monoklonal» angir karakteren til antistoffet som fås fra en stort sett homogen populasjon av antistoffer, og skal ikke tolkes slik at det krever at antistoffet produseres med en bestemt metode. For eksempel kan de monoklonale antistoffene som skal anvendes ifølge den foreliggende oppfinnelsen fremstilles ved en rekke metoder, inkludert f.eks. hybridommetoden (f.eks. Kohler and Milstein., Nature, 26:49-97 (197); Hongo et al., Hybridoma, 14 (3): (199), Harlow et al., Antibodies: A Laboratory Manual, (Cold Spring Harbor Laboratory Press, 2. utg. 1988); Hammerling et al., i: Monoclonal Antibodies and T-Cell Hybridomas (Elsevier, N.Y., 1981)), rekombinante DNA-metoder (se f.eks. U.S. patent nr ), bakteriofagdisplayteknologier (se f.eks. Clackson et al., Nature, 32: (1991); Marks et al., J. Mol. Biol. 222: (1992); Sidhu et al., J. Mol. Biol. 338(2): (2004); Lee et al., J. Mol. Biol. 340(): (2004); Fellouse, Proc. Natl. Acad. Sci. USA 1 (34): (2004); og Lee et al., J. Immunol. Methods 284(1-2): (2004), og teknologier for å produsere humane eller menneskelignende antistoffer hos dyr som har deler eller hele den menneskelige immunoglobulinlokusen eller gener som koder for humane immunoglobulinsekvenser (se f.eks. WO 1998/24893; WO 1996/34096; WO 1996/3373; WO 1991/741; Jakobovits et al., Proc. Natl. Acad. Sci. USA 90: 21 (1993); Jakobovits et al., Nature 362: 2 28 (1993); Bruggemann et al., Year in Immunol. 7:33 (1993); U.S. Patent nr ; 4 806; 69 82; ; ; og ; Marks et al., Biotechnology : (1992); Lonberg et al., Nature 368: (1994); Morrison, Nature 368: (1994); Fishwild et al., Nature Biotechnol. 14: (1996); Neuberger, Nature Biotechnol. 14: 826 (1996); og Lonberg and Huszar, Intern. Rev. Immunol. 13: 6 93 (199). Uttrykket «nakent antistoff» står for et antistoff som ikke er konjugert til en cytotoksisk gruppe eller radioaktiv merking. 3 Uttrykkene «fullengdeantistoff», «intakt antistoff» eller «helt antistoff» anvendes om hverandre for å betegne et antistoff i sin stort sett intakte form, i motsetning til et antistoffragment. Nærmere bestemt inkluderer de hele antistoffene
30 28 antistoffer med tung og lett kjede, inkludert en Fc-region. De konstante domenene kan være naturlige sekvenskonstante domener (f.eks. humane naturlige sekvenskonstante domener) eller aminosyresekvensvarianter av disse. I noen tilfeller kan det intakte antistoffet ha én eller flere effektorfunksjoner. Et «antistoffragment» omfatter en del av et intakt antistoff, fortrinnsvis antigenbindingen og/eller den variable regionen av det intakte antistoffet. Eksempler på antistoffragmenter inkluderer Fab-, Fab'-, F(ab') 2 - og Fvfragmenter; dialegemer; lineære antistoffer (se U.S. patent , eksempel 2; Zapata et al., Protein Eng. 8(): 7 62 [199]); enkeltkjedeantistoffmolekyler og flerspesifikke antistoffer dannet av antistoffragmenter Papaindigerering av antistoffer som produserte to identiske antigenbindende fragmenter, kalt «Fab»-fragmenter, og et rest-«fc»-fragment, en betegnelse som gjenspeiler at det har evnen til å krystallisere lett. Fab-fragmentet består av en hel L-kjede sammen med domenet for den variable regionen til H-kjeden (V H ), og det første konstante domenet til en tung kjede (C H 1). Hvert Fab-fragment er enverdig med hensyn til antigenbinding, dvs. at det har et enkelt antigenbindingssete. Pepsinbehandling av et antistoff gir et enkelt stort F(ab') 2 -fragment, som omtrent tilsvarer to disulfidbundne Fab-fragmenter som har forskjellig antigenbindende aktivitet, og fremdeles er i stand til å kryssbinde antigen. Fab'-fragmenter skiller seg fra Fab-fragmenter ved at de har noen få flere aminosyrerester i karboksyenden til C H 1-domenet, inkludert én eller flere cysteiner fra antistoffhengselregionen. Fab'- SH er betegnelsen i dette dokumentet for Fab' der cysteinresten(e) til de konstante domenene har en fri tiolgruppe. F(ab') 2 -antistoffragmenter ble opprinnelig produsert som par av Fab'-fragmenter som har hengselcysteiner mellom seg. Andre kjemiske koblinger av antistoffragmenter er også kjent. 30 Fc-fragmentet omfatter karboksyendedelene av begge H-kjedene holdt sammen med disulfider. Effektorfunksjonene til antistoffene bestemmes av sekvensene i Fc-regionen, som også gjenkjennes av Fc-reseptorer (FcR) som finnes på visse celletyper. 3 «Fv» er det minste antistoffragmentet som inneholder en/et fullstendig antigengjenkjennelses- og bindingssete. Dette fragmentet består av en dimer av det variable regiondomenet fra en tung og en lett kjede i tett, ikke-kovalent forbindelse. Fra foldingen av disse to domenene utgår det seks hypervariable
31 29 løkker (3 løkker fra hver av H- og L-kjeden) som bidrar med aminosyrerestene for antigenbinding og gir antigenbindende spesifisitet til antistoffet. Imidlertid har til og med et enkelt variabelt domene (eller halvparten av et Fv som bare omfatter tre HVR-er spesifikke for et antigen) evnen til å gjenkjenne og binde antigen, men med lavere affinitet enn hele bindingssetet. «Enkeltkjede-Fv», også forkortet til «sfv» eller «scfv» er antistoffragmenter som omfatter VH- og VL-antistoffdomenene koblet til en enkelt polypeptidkjede. Fortrinnsvis omfatter sfv-polypeptidet også et polypeptidbindeledd mellom V H - og V L -domenene som gjør det mulig for sfv å danne den ønskede strukturen for antigenbinding. En gjennomgang av sfv finnes i Pluckthun in The Pharmacology of Monoclonal Antibodies, bd. 113, Rosenburg and Moore eds., Springer-Verlag, New York, s (1994) «Funksjonelle fragmenter» av antistoffene ifølge oppfinnelsen omfatter en del av et intakt antistoff, vanligvis inkludert antigenbindingsregionen eller den variable regionen til det intakte antistoffet eller F-regionen til et antistoff som beholder eller har modifisert FcR-bindingsevne. Eksempler på antistoffragmenter inkluderer lineært antistoff, enkeltkjedeantistoffmolekyler og flerspesifikke antistoffer dannet av antistoffragmenter Betegnelsen «dialegemer» står for små antistoffragmenter som fremstilles ved å konstruere sfv-fragmenter (se foregående avsnitt) med korte bindeledd (omtrent ) rester) mellom V H - og V L -domenene slik V-domenene pares mellom kjedene og ikke innen kjedene slik at det blir et toverdig fragment, dvs. et fragment som har to antigenbindingsseter. Bispesifikke dialegemer er heterodimerer av to «overkryssende» sfv-fragmenter der V H - og V L -domenene til de to antistoffene befinner seg på forskjellige polypeptidkjeder. Dialegemer er beskrevet i større detalj i f.eks. EP ; WO 93/11161; Hollinger et al., Proc. Natl. Acad. Sci. USA 90: (1993). 3 De monoklonale antistoffene i dette dokumentet inkluderer spesifikt «kimære» antistoffer (immunoglobuliner) der en del av den tunge og/eller lette kjeden er identisk med eller homolog med tilsvarende sekvenser i antistoffer avledet fra en bestemt art, eller som tilhører en bestemt antistoffklasse eller underklasse, mens resten av kjeden(e) er identisk(e) med eller homolog(e) med tilsvarende sekvenser i antistoffer avledet fra en annen art eller som tilhører en annen
32 30 antistoffklasse eller underklasse, så vel som fragmenter av slike antistoffer, så lenge de viser den ønskede biologiske aktiviteten (U.S. patent nr ; Morrison et al., Proc. Natl. Acad. Sci. USA, 81: (1984)). Kimære antistoffer av interesse i dette dokumentet inkluderer PRIMATIZED antistoffer, der den antigenbindende regionen til antistoffet avledes fra et antistoff som produseres av f.eks. immuniserende makaker med et antigen av interesse. Slik det anvendes i dette dokumentet står «humanisert antistoff» for en undergruppe av «kimære antistoffer» «Humaniserte» former av ikke-humane (f.eks. murine) antistoffer er kimære antistoffer som inneholder en minimal sekvens avledet fra ikke-humant immunoglobulin. I en utførelsesform er et humanisert antistoff et humant immunoglobulin (mottakerantistoff) der aminosyrerester fra en HVR fra mottakeren byttes ut med rester fra en HVR av en ikke-human art (donorantistoff) som f.eks. mus, rotte, kanin eller ikke-human primat med den ønskede spesifisiteten, affiniteten og/eller kapasiteten. I noen tilfeller byttes FR-rester i humant immunoglobulin ut med tilsvarende ikke-humane rester. Videre kan de humaniserte antistoffene omfatte rester som ikke finnes i mottakerens antistoff eller i donorantistoffet. Disse modifikasjonene kan utføres for å forfine antistoffytelsen ytterligere, f.eks. bindingsaffiniteten. Generelt vil et humanisert antistoff omfatte stort sett alt av minst ett, og typisk to variable domener, der alle eller i det vesentlige alle de hypervariable løkkene tilsvarer løkkene til en ikke-human immunoglobulinsekvens, og hele eller en vesentlig del av FR-regionene er fra en human immunoglobulinsekvens, selv om FR-regionene kan inkludere én eller flere individuelle FR-restsubstitusjoner som forbedrer antistoffytelsen, f.eks. bindingsaffinitet, isomerisering, immunogenisitet osv. Antallet av disse aminosyresubstitusjonene i FR-en er vanligvis ikke mer enn 6 i H-kjeden, og ikke mer enn 3 i L-kjeden. Det humaniserte antistoffet vil eventuelt også omfatte minst en del av en konstant immunoglobulinregion (Fc), vanligvis fra et humant immunoglobulin. Flere opplysninger finnes f.eks. i Jones et al., Nature 321:22 2 (1986); Riechmann et al., Nature 332: (1988); og Presta, Curr. Op. Struct. Biol. 2:93 96 (1992). Se også f.eks. Vaswani og Hamilton, Ann. Allergy, Asthma & Immunol. 1: 11 (1998); Harris, Biochem. Soc. Transactions 23:3 38 (199); Hurle og Gross, Curr. Op. Biotech. : (1994); og U.S. Pat. nr og
33 Et «humant antistoff» er et som besitter en aminosyresekvens som tilsvarer aminosyresekvensen til et antistoff som produseres av et menneske og/eller er blitt laget ved hjelp av en hvilken som helst av metodene for å lage humane antistoffer som er offentliggjort i dette dokumentet. Denne definisjonen på et humant antistoff utelukker spesifikt et humanisert antistoff som omfatter ikkehumane antigenbindende rester. Humane antistoffer kan produseres ved hjelp av forskjellige metoder som er kjent i faget, inkludert bakteriofagdisplaybibliotek. Hoogenboom and Winter, J. Mol. Biol., 227:381 (1991); Marks et al., J. Mol. Biol., 222:81 (1991). Også metodene som er beskrevet i Cole et al., Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, s. 77 (198); Boerner et al., J. Immunol., 147(1):86 9 (1991) er tilgjengelig for fremstilling av humane monoklonale antistoffer er. Se også van Dijk og van de Winkel, Curro Opin. Pharmacol., : (2001). Humane antistoffer kan fremstilles ved å administrere antigenet til et transgent dyr som er blitt modifisert for å produsere slike antistoffer som respons på antigenutfordring, men som de endogene lokusene er deaktivert for, f.eks. immuniserte xenomus (se f.eks. U.S. Pat. nr og 6 84 angående XENOMOUSE -teknologi). Se også f.eks. Li et al., Proc. Natl. Acad. Sci. USA, 3: (2006) når det gjelder humane antistoffer som dannes av humane B-celler ved en hybridomteknologi. Når uttrykkene «hypervariabel region», «HVR» eller «HV» anvendes i dette dokumentet, henviser de til regioner av et antistoffvariabelt domene som er hypervariable i sekvens og/eller danner strukturelt definerte løkker. Antistoffer omfatter generelt seks HVR; tre i VH (H1, H2, H3), og tre i VL-en (L1, L2, L3). I naturlige antistoffer viser H3 og L3 det største mangfoldet av de seks HVRene, og særlig antas H3 å spille en unik rolle i forbindelse med å gi antistoffene fin spesifisitet. Se f.eks. Xu et al. Immnunity 13:37 4 (2000); Johnson og Wu i Methods in Molecular Biology 248:1 2 (Lo, red., Human Press, Totowa, NJ, 2003)). Faktisk er naturlig forekommende kameldyrantistoffer som bare består av en tung kjede, funksjonelle og stabile i fravær av en lett kjede. Se f.eks. Hamers-Casterman et al., Nature 363: (1993) og Sheriff et al., Nature Struck. Biol. 3: (1996). 3 En rekke HVR-avgrensninger er i anvendelse og omfattes i dette dokumentet. HVR-ene som er Kabat-komplementaritetsbestemmende regioner (CDR-er) bygger på sekvensvariabilitet og er de som anvendes hyppigst (Kabat et al., ovenfor). Chothia henviser i stedet til plasseringen av de strukturelle løkkene
34 32 (Chothia and Lesk J. Mol. Biol. 196: (1987)). AbM HVR representerer et kompromiss mellom Kabats CDR og Chothias strukturelle løkker, og anvendes av Oxford Moleculars AbM antistoffmodelleringsprogramvare. «Kontakt»-HVR bygger på en analyse av de tilgjengelige komplekse krystallstrukturene. Restene fra hver av disse HVR-ene er omtalt nedenfor. Løkke Kabat AbM Chothia Kontakt L1 L24 L34 L24 L34 L26 L34 L30 L36 L2 L0 L6 L0 L6 L0 L6 L46 L L3 L89 L97 L89 L97 L91 L96 L89 L96 H1 H31 H3B H26 H3B H26 H32 H30 H3B (Kabat-nummerering) H1 H31 H3 H26 H3 H26 H32 H30 H3 (Chothia-nummerering) H2 H0 H6 H0 H8 H3 H6 H47 H8 H3 H9 H2 H9 H2 H9 H2 H93 H1 HVR kan omfatte «utvidede HVR» som følger: eller (L1), 46 6 eller 0 6 (L2) og eller (L3) i VL og 26 3 (H1), 0 6 eller 47 6 (en foretrukket utførelsesform) (H2) og 93 2 (H3) i VH. De variable domenerestene er nummerert ifølge Kabat et al., ovenfor, for hver av disse utvidede HVR-definisjonene. 1 «Ramme»- eller «FR»-rester er de variabelt domene-restene som ikke er HVRrester, som definert i dette dokumentet. 20 Uttrykket «nummerering av variabelt domene-rester som i Kabat» eller «posisjonsnummerering for aminosyrer som i Kabat», og varianter av disse henviser til nummereringssystemet som anvendes for variable domener i den tunge eller lette kjeden i sammenstillingen av antistoffer i Kabat et al., ovenfor. Ved hjelp av dette nummereringssystemet kan den faktiske lineære aminosyresekvensen inneholde færre eller flere aminosyrer som tilsvarer en forkorting av, eller innsetting i en FR eller HVR i det variable domenet. Et variabelt domene i den tunge kjeden kan f.eks. inkludere å sette inn en enkelt aminosyre (rest 2a ifølge Kabat) etter
35 rest 2 i H2, og innsatte rester (f.eks. restene 82a, 82b, og 82c osv. ifølge Kabat) etter FR-rest 82 i den tunge kjeden. Kabat-nummereringen av restene kan bestemmes for et gitt antistoff ved sammenlikning med en «standard» Kabatnummerert sekvens ved homologiregioner av sekvensen til antistoffet. En «akseptorhuman ramme» står i dette dokumentet for en ramme som omfatter aminosyresekvensen til en VL- eller VH-ramme avledet fra en human immunoglobulinramme eller en human konsensusramme. En akseptorhuman ramme «avledet fra» en human immunoglobulinramme eller en human konsensusramme kan omfatte den samme aminosyresekvensen eller den kan inneholde aminosyresekvensendringer som eksisterte fra før. I noen utførelsesformer er antallet aminosyreendringer som eksisterte fra før eller mindre, 9 eller mindre, 8 eller mindre, 7 eller mindre, 6 eller mindre, eller mindre, 4 eller mindre, 3 eller mindre, 2 eller mindre eller mindre. En «human konsensusramme» er en ramme som representerer de hyppigst forekommende aminosyrerestene i et utvalg av humane immunoglobulin VLeller VH-rammesekvenser. Generelt velges humane immunoglobulin-vl- eller - VH-sekvenser fra en undergruppe av variable domenesekvenser. Vanligvis er undergruppen av sekvenser en undergruppe som i Kabat et al., Sequences of Proteins of Immunological Interest,. utg. Public Health Service, National Institutes of Health, Bethesda, MD (1991). I en utførelsesform for VL er undergruppen undergruppe kappa I som i Kabat et al., ovenfor. I en utførelsesform for VH er undergruppen undergruppe III som i Kabat et al., ovenfor. 30 En «konsensusramme fra VH-undergruppe II» omfatter konsensussekvensen fra aminosyresekvensene i variabel tung undergruppe III i Kabat et al., ovenfor. I én utførelsesform omfatter aminosyresekvensen til konsensusrammen i VHundergruppe III minst en del av eller alle de følgende sekvensene: EVQLVESGGGLVQPGGSLRLSCAAS (H1)(SEQ ID NO:), WVRQAPGKGLEWVA (SEQ ID NO:6)(H2), RFTISRDDSKNTLYLQMNSLRAEDTAVYYCAR (SEQ ID NO:7)(H3), WGQGTLVTVSS (SEQ ID NO:8)(H4). 3 En «konsensusramme i VL-undergruppe I» omfatter konsensussekvensen fra aminosyresekvensene i den variable lette kappa-undergruppe I i Kabat et al., ovenfor. I én utførelsesform omfatter aminosyresekvensen til konsensusrammen
36 34 i VH-undergruppe I minst en del av eller alle de følgende sekvensene: DIQMTQSPSSLSASVGDRVTITC (SEQ ID NO:9)(L1), WYQQKPGKAPKLLIY (SEQ ID NO:60)(L2), GVPSRFSGSGSGTDFTLTISSLQPEDFATYYC (SEQ ID NO:61)(L3), FGQGTKVEIKR (SEQ ID NO:62)(L4). En «aminosyremodifikasjon» i en bestemt posisjon, f.eks. i Fc-regionen, står for substitusjon eller sletting av den spesifiserte resten, eller innsetting av minst en aminosyrerest inntil den spesifiserte resten. Innsetting «inntil» en spesifisert rest betyr innsetting innen en til to rester fra denne. Innsettingen kan være i N-enden eller C-enden av den spesifiserte resten. I dette dokumentet er den foretrukne aminosyremodifikasjonen en substitusjon Et «affinitetsmodnet» antistoff er et antistoff med én eller flere forandringer i én eller flere HVR i den som fører til en forbedring av affiniteten til antistoffet for antigen, sammenlignet med et opphavsantistoff som ikke besitter denne/disse endringen(e). I én utførelsesform har et affinitetsmodnet antistoff nanomolar eller til og med pikomolar affinitet for målantigenet. Affinitetsmodnede antistoffer produseres ved prosedyrer som er kjent i faget. F.eks. beskriver Marks et al., Biotechnology : (1992) affinitetsmodning ved omstokking av VH- og VL-domenet. Tilfeldig mutagenese av HVR- og/eller rammerester beskrives f.eks. av: Barbas et al. Proc Nat. A cad. Sci. USA 91: (1994); Schier et al. Gene 169:147 1 (199); Yelton et al. J. Immunol. 1: (199); Jackson et al., J. Immunol. 14(7):33 9 (199); og Hawkins et al, J. Mol. Biol. 226: (1992). Et antistoff som «spesifikt binder» eller er «spesifikt for» et bestemt polypeptid eller en epitop av et bestemt polypeptid er et antistoff som binder det aktuelle polypeptidet eller epitopen på et bestemt polypeptid uten å binde noe annet polypeptid eller noen annen polypeptidepitop i betydelig grad. F.eks. er de M1'- spesifikke antistoffene ifølge den foreliggende oppfinnelsen spesifikke for det M1'-ekstracellulære segmentet av IgE som finnes på IgE-bundet på B-celler, men som ikke er til stede på utskilt IgE. 3 Et «blokkerende» antistoff eller et «antagonist»-antistoff er et antistoff som hemmer eller reduserer en biologisk aktivitet hos antigenet det binder. I noen utførelsesformer hemmer blokkerende antistoffer eller antagonistantistoffer den biologiske aktiviteten til antigenet i betydelig grad eller fullstendig. De apoptotiske
37 3 antistoffene mot IgE ifølge oppfinnelsen blokkerer aktiviteten til IgE med å formidle immunrespons på en slik måte at det desimerer både fritt og totalt serum-ige. Antistoffer som «utløser apoptose» eller er «apoptotiske» er antistoffer som utløser programmert celledød ifølge bestemmelser ved standard apoptoseanalyser, som binding av annexin V, fragmentering av DNA, cellekrymping, utvidelse av det endoplasmiske retikulum, cellefragmentering og/eller dannelse av membranvesikler (kalt apoptotiske legemer). For eksempel kan den apoptotiske aktiviteten til antistoffene mot IgE i den foreliggende oppfinnelsen vises ved å farge cellene som har overflatebundet IgE med annexin V. 1 Uttrykket «totalt serum-ige» står for den totale mengden av IgE som er til stede i en prøve, inkludert både fritt eller ubundet IgE, i tillegg til IgE som har dannet kompleks med en bindingspartner (f.eks. antistoff mot IgE, IgE-bærende B- celle). «Fritt serum-ige» står for IgE som ikke er bundet til en bindingspartner Uttrykket «allergenspesifikk IgE» står for IgE som er spesifikt for et spesielt antigen som stammer fra en første eksponering for allergen i en prosess kjent som allergisensibilisering, og som binder overflaten av mastceller og basofiler, og som kan føre til aktivering av mastceller og basofiler ved senere eksponering for det samme allergenet. Flere mitogene faktorer i virus (f.eks. cytomegalovirus CMV), bakterier (f.eks. Staphylococcus), helminter (f.eks. Ascaris, Schistosoma) og adjuvansfaktorer i luftforurensning (f.eks. sigarettrøyk og dieseleksos) stimulerer produksjonen av IgE-molekyler uten å utløse en allergenspesifikk IgE-sensibilisering. Fordi IgE-konsentrasjoner kan øke på en måte som ikke nødvendigvis disponerer verten til å bli mer mottagelig for en IgE-formidlet forstyrrelse, anvendes det derfor av og til allergenspesifikke IgE-konsentrasjoner i kliniske vurderinger Uttrykket «spesifikk desimering av IgE-produserende B-celler» betyr evnen til å spesifikt redusere populasjonen av eller effektiviteten av B-celler som spesifikt skiller ut IgE (dvs. plasmaceller), men som ikke i vesentlig grad påvirker populasjonen eller effektiviteten til B-celler som skiller ut andre immunoglobuliner, som f.eks. IgG1, IgG2, IgG3, IgG4, IgA og IgM. Uttrykket «fast fase» beskriver en ikke-vandig matriks som antistoffet ifølge den foreliggende oppfinnelsen kan klebe seg til. Eksempler på faste faser som omfattes i
38 36 dette dokumentet inkluderer slike som er laget helt eller delvis av glass (f.eks. glass med kontrollert porediameter), polysakkarider (f.eks. agarose), polyakrylamider, polystyren, polyvinylalkohol og silikoner. I visse utførelsesformer, avhengig av sammenhengen, kan den faste fasen omfatte brønnen på en analyseplate, i andre er det en rensekolonne (f.eks. en affinitetskromatografikolonne). Dette begrepet inkluderer også en diskontinuerlig fast fase av adskilte partikler, som f.eks. slike som er beskrevet i U.S. patent nr Antistoff-«effektorfunksjoner» står for de biologiske aktivitetene som kan tilskrives Fc-regionen (en Fc-region med naturlig sekvens eller Fc-region med variantaminosyresekvens) til et antistoff, og varierer med antistoffisotypen. Eksempler på antistoffeffektorfunksjoner inkluderer: C1q-binding og komplementavhengig cytotoksisitet, Fc-reseptorbinding, antistoffavhengig celleformidlet cytotoksisitet (ADCC), fagocytose, nedregulering av celleoverflatereseptorer (f.eks. B-cellereseptorer) og B-celleaktivering «Antistoffavhengig celleformidlet cytotoksisitet» eller ADCC står for en form for cytotoksisitet der utskilt Ig bundet til Fc reseptorer (FcR-er) som finnes på visse cytotoksiske celler (f.eks. naturlige dreperceller (NK-celler), nøytrofiler og makrofager) gjør det mulig for disse cytotoksiske effektorcellene å binde en antigenbærende målcelle spesifikt og deretter drepe målcellen med cytotoksiner. Antistoffene «armerer» de cytotoksiske cellene og er nødvendige for å drepe målcellen ved denne mekanismen. De primære cellene for formidling av ADCC, NKcellene, uttrykker bare FcγRIII, mens monocytter uttrykker FcγRI, FcγRII og FcγRIII. Fc-uttrykkingen på hematopoetiske celler er oppsummert i tabell 3 på side 464 av Ravetch and Kinet, Annu. Rev. Immunol. 9: (1991). For å vurdere ADCC-aktiviteten til et molekyl av interesse kan det utføres en ADCC-analyse in vitro, som den som er beskrevet i U.S. patent nr eller Nyttige effektorceller for slike analyser inkluderer perifere mononukleære blodceller (PBMC) og naturlige dreperceller (NK-celler). Alternativt, eller i tillegg, kan ADCCaktiviteten til molekylet av interesse vurderes in vivo, f.eks. i en dyremodell som den som er beskrevet i Clynes et al., PNAS USA 9:62 66 (1998). 3 Med mindre det er angitt noe annet i dette dokumentet, er nummereringen av restene i en tung immunoglobulinkjede fra EU-indeksen som i Kabat et al., ovenfor. «EU-indeksen som i Kabat» står for nummereringen av aminosyrerester i det humane IgG1 EU-antistoffet.
39 37 1 Betegnelsen «Fc-region» anvendes i dette dokumentet for å definere en C- enderegion i en tung immunoglobulinkjede, inkludert Fc-regioner med naturlig sekvens og variant-fc-regioner. Selv om grensene til Fc-regionen i en tung immunoglobulinkjede kan variere, defineres Fc-regionen i den tunge kjeden av humant IgG vanligvis slik at det strekker seg fra en aminosyrerest i posisjon Cys226, eller fra Pro230, til karboksylenden av regionen. Lysinet i C-enden (rest 447 etter EU-nummereringssystemet) av Fc-regionen kan fjernes f.eks. under produksjon eller rensing av antistoffet, eller ved rekombinant konstruering av nukleinsyren som koder for den tunge kjeden til antistoffet. Følgelig kan en sammensetning av intakte antistoffer omfatte antistoffpopulasjoner der alle K447-restene er fjernet, antistoffpopulasjoner der ingen av K447-restene er fjernet, og antistoffpopulasjoner som har en blanding av antistoffer med og uten K447-resten. Egnede Fc-regioner med naturlig sekvens til anvendelse i antistoffene ifølge oppfinnelsen inkluderer human IgG1, IgG2, IgG3 og IgG «Fc-reseptor» eller «FcR» beskriver en reseptor som bindes til Fc-regionen av et antistoff. Den foretrukne FcR-en er en human FcR med naturlig sekvens. Videre er en foretrukket FcR en FcR som binder et IgG-antistoff (en gammareseptor) og inkluderer reseptorer for FcγRI-, FcγRII- og FcγRIII-underklassene, inkludert alleliske varianter og alternativt spleisede former av disse reseptorene, FcγRIIreseptorer inkluderer FcγRIIA (en «aktiverende reseptor») og FcγRIIB (en «hemmende reseptor»), som har tilsvarende aminosyresekvenser som skiller seg fra hverandre først og fremst i de cytoplasmiske domenene. Aktiverende reseptor FcγRIIA inneholder et immunreseptortyrosinbasert aktiveringsmotiv (ITAM) i det cytoplasmiske domenet sitt. Hemmereseptoren FcγRIIB inneholder en immunreseptortyrosinbasert hemmerest (ITIM) i det cytoplasmiske domenet sitt. (se M. Daëron, Annu. Rev. Immunol. 1: (1997). FcR-ene er gjennomgått i Ravetch and Kinet, Annu. Rev. Immunol. 9: (1991); Capel et al., Immunomethods 4: 2 34 (1994); og de Haas et al., J. Lab. Clin. Med. 126: (199). Andre FcR, inkludert de som vil bli identifisert i fremtiden, omfattes også av betegnelsen «FcR» i dette dokumentet. 3 Betegnelsen «Fc-reseptor» eller «FcR» inkluderer også den neonatale reseptoren, FcRn, som er ansvarlig for overføring av morens IgG til fosteret. Guyer et al., J. Immunol. 117: 87 (1976) og Kim et al., J. Immunol. 24: 249 (1994). Metoder for måling av binding til FcRn er kjent (se f.eks. Ghetie and
40 38 Ward, Immunol. Today 18: (12): 92 8 (1997); Ghetie et al., Nature Biotechnology 1 (7): (1997); Hinton et al., J. Biol. Chem. 279 (8): (2004); WO 2004/92219 (Hinton et al.). Binding til FcRn in vivo og serumhalveringstiden til humane FcRnbindingspolypeptider med høy affinitet kan analyseres f.eks. i transgene mus eller transfekterte humane cellestammer som uttrykker human FcRn, eller i primater som polypeptidene med en variant-fc-region er administrert til. WO 2004/42072 (Presta) beskriver antistoffvarianter med forbedret eller redusert binding til FcRer. Se også f.eks. Shields et al., J. Biol. Chem. 9(2): (2001). 1 «Humane effektorceller» er leukocytter som uttrykker én eller flere FcR og utfører effektorfunksjoner. Fortrinnsvis uttrykker cellene i det minste FcγRIII og utfører ADCC-effektorfunksjon. Eksempler på humane leukocytter som formidler ADCC inkluderer mononukleære celler fra perifert blod (PBMC), naturlige dreperceller (NKceller), monocytter, cytotoksiske T-celler og nøytrofiler, der PBMC og MNK-celler er foretrukket. Effektorcellene kan isoleres fra en naturlig kilde, f.eks. blod «Komplementavhengig cytotoksisitet» eller «CDC» står for lyseringen av en målcelle i nærvær av komplement. Aktivering av den klassiske komplementreaksjonsveien starter med binding av den første delen av komplementsystemet (C1q) til antistoffer (av den egnede underklassen) som er bundet til det tilsvarende antigenet. For å vurdere komplementaktiveringen kan det utføres en CDC-analyse, f.eks. som beskrevet i Gazzano-Santoro et al., J. Immunol. Methods 202: 163 (1996). 30 Polypeptidvarianter med endrede aminosyresekvenser i Fc-regionen og økt eller redusert C1q-bindingsevne er beskrevet i US patent nr B1 og WO99/1642. Innholdet i disse patentpublikasjonene er innført spesifikt i dette dokumentet ved referanse. Se også Idusogie et al. J. Immunol. 164: (2000). 3 N-glykosyleringssetet i IgG er ved Asn297 i CH2-domenet. Den foreliggende oppfinnelsen tilveiebringer også sammensetninger av et CD20-bindende humanisert antistoff som har en Fc-region, der omtrent 80 til 0 % (og fortrinnsvis omtrent 90 til 99 %) av antistoffet i sammensetningen omfatter en karbohydratstruktur med moden kjerne som mangler fukose, festet til Fc-regionen av glykoproteinet. Det er
41 39 demonstrert i dette dokumentet at slike sammensetninger viser en overraskende forbedring når det gjelder binding til FcγRIIIA (F18), som ikke er så effektiv som FcγRIIIA (V18) i samspill med humant IgG. Dermed forventes sammensetningene i dette dokumentet å være overlegne i forhold til tidligere beskrevne antistoffsammensetninger mot CD20, spesielt for behandling av humane pasienter som uttrykker FcγRIIIA (F18). FcγRIIIA (F18) er mer vanlig enn FcγRIIIA (V18) hos normale, friske afroamerikanere og kaukasoide. Se Lehrnbecher et al. Blood 94:4220 (1999). Den foreliggende søknaden viser også den synergistiske økningen i FcγRIII-binding og/eller ADCC-funksjon som følger av å kombinere glykosyleringsvariasjonene i dette dokumentet med aminosyresekvensmodifikasjon(er) i Fc-regionen til glykoproteinet «Bindingsaffinitet» står generelt for styrken på summen av ikke-kovalente interaksjoner mellom et enkelt bindingssete på et molekyl (f.eks. et antistoff) og bindingspartneren (f.eks. et antigen). Med mindre det er angitt noe annet står «bindingsaffinitet» slik det anvendes i dette dokumentet for den iboende bindingsaffiniteten som gjenspeiler en 1:1-interaksjon mellom medlemmer av et bindingspar (f.eks. antistoff og antigen). Affiniteten til et molekyl X for partneren sin Y kan generelt representeres ved dissosiasjonskonstanten (K d ). Affiniteten kan måles ved vanlige metoder som er kjent i faget, inkludert de som beskrives i dette dokumentet. Lavaffinitetsantistoffer binder generelt antigen langsomt og er tilbøyelig til å dissosiere lett, mens antistoffer med høy affinitet generelt binder antigen raskere og er tilbøyelig til å forbli bundet lengre. Det er kjent en rekke metoder for å måle bindingsaffinitet innen faget, og alle kan anvendes til formålene i den foreliggende oppfinnelsen. Spesifikke illustrerende og eksempelvise utførelsesformer for å måle bindingsaffinitet er beskrevet i det følgende I én utførelsesform måles «K d» eller «K d -verdien» ifølge denne oppfinnelsen ved en radioaktivt merket antigenbindingsanalyse (RIA) utført med Fab-versjonen til et antistoff av interesse og antigenet til denne som beskrevet i den følgende analysen. Løsningsbindingsaffiniteten til Fab-er for antigen måles ved å innstille likevekt mellom Fab og en minimal konsentrasjon av ( 12 I)-merket antigen i nærvær av en titreringsserie med umerket antigen, og deretter fange opp bundet antigen med en plate belagt med antistoff mot Fab (se f.eks. Chen et al., J. Mol. Biol. 293: (1999)). For å etablere forholdene for analysen dekkes mikrotiterplater (DYNEX Technologies, Inc.) over natten med µg/ml av et innfangingsantistoff mot Fab (Cappel Labs) i 0 mm natriumkarbonat (ph 9,6), og blokkeres deretter med 2 %
42 40 (vekt/volum) bovint serumalbumin i PBS i to til fem timer ved romtemperatur (omtrent 23 C). På en ikke-adsorberende plate (Nunc #269620) blandes 0 pm eller 26 pm [ 12 I]-antigen med seriefortynninger av et Fab av interesse (f.eks. i overensstemmelse med vurderingen av anti-vegf-antistoffet, Fab-12, i Presta et al., Cancer Res. 7: (1997)). Fab-et av interesse inkuberes deretter over natten; inkuberingen kan imidlertid fortsette i en lengre periode (f.eks. omtrent 6 timer) for å sikre at det er oppnådd likevekt. Deretter overføres blandingene til opptaksplaten for inkubering ved romtemperatur (f.eks. en time). Løsningen blir deretter fjernet og platen vasket åtte ganger med 0,1 % TWEEN-20 surfaktant i PBS. Når platene har tørket blir µl/brønn scintilleringsmiddel (MICROSCINT- 20 ; Packard) tilsatt, og platene telles på en TOPCOUNT gammateller (Packard) i ti minutter. Konsentrasjonene av hver Fab som gir mindre enn eller lik 20 % maksimal binding blir valgt for å anvendes i konkurrerende bindingsanalyser Ifølge en annen utførelsesform måles K d ved overflateplasmonresonansanalyser med et BIACORE eller et BIACORE instrument (BIAcore, Inc., Piscataway, NJ) ved 2 C med immobiliserte antigen CM-brikker og ~ responsenheter (RU). Kort fortalt ble karboksymetylerte dekstranbiosensorbrikker (CM, Biacore Inc.) aktivert med N-etyl-N'-(3-dimetylaminopropyl)- karbodiimidhydroklorid (EDC) og N-hydroksysuksinimid (NHS) etter leverandørens instruksjoner. Antigenet ble fortynnet med mm natriumacetat, ph 4,8, til µg/ml (~0,2 µm) før injeksjon med en strømningshastighet på µl/minutt for å oppnå omtrent responsheter (RU) av koblet protein. Etter injeksjonen av antigenet ble det injisert 1 M etanolamin for å blokkere ureagerte grupper. For kinetiske målinger ble seriefortynninger av Fab (0,78 nm til 00 nm) injisert to ganger i PBS med 0,0 % TWEEN 20 -surfaktant (PBST) ved 2 C og en strømningshastighet på omtrent 2 µl/min. Assosiasjonshastigheter (k on ) og dissosiasjonshastigheter (k off ) ble beregnet ved å anvende en enkel én-til-én Langmuir-bindingsmodell (BIAcore Evaluation Software versjon 3.2) ved samtidig montering av assosierings- og dissosieringssensorgrammene. Denne likevektdissosiasjonskonstanten (K d ) ble beregnet som forholdet k off :k on. Se f.eks. Chen et al., J. Mol. Biol. 293: (1999). Hvis on-hastigheten overskrider 6 M -1 s -1 ved overflateplasmonresonansanalysen ovenfor kan on-raten bestemmes ved en fluorescensdempemetode der man måler økningen eller reduksjonen i fluorescensemisjonsintensiteten (eksitasjon = 29 nm, emisjon = 340 nm, 16 nm båndpass) ved 2 C av et 20 nm anti-antigenantistoff (Fab-form) i PBS, ph 7,2, i nærvær av økende konsentrasjoner av antigen ifølge spektrometermålinger, f.eks.
43 41 et stoppstrømutstyrt spektrofotometer (Aviv Instruments) eller et SLM-AMINCO spektrofotometer av 8000-serien (ThermoSpectronic) med en omrørt kyvette. En «on-rate», «assosiasjonshastighet» eller «k on» ifølge denne oppfinnelsen kan også bestemmes som beskrevet ovenfor med et BIACORE 2000-system eller et BIACORE 3000-system (BIAcore, Inc., Piscataway, NJ). 1 Uttrykket «vesentlig redusert» eller «vesentlig forskjellig», slik det anvendes i dette dokumentet, betegner en tilstrekkelig høy grad av forskjell mellom to numeriske verdier (generelt forbundet med et molekyl, og den andre forbundet med et referanse-/sammenligningsmolekyl), slik at en fagperson vil vurdere forskjellen mellom de to verdiene som statistisk signifikante i sammenheng med det biologiske karaktertrekket som måles ved verdiene (f.eks. K d -verdier). Forskjellen mellom de to verdiene er f.eks. større enn omtrent %, større enn omtrent 20 %, større enn omtrent 30 %, større enn omtrent 40 % og/eller større enn omtrent 0 % som funksjon av verdien til referanse-/sammenligningsmolekylet Uttrykkene «i det vesentlige lik» eller «i det vesentlige den samme», slik de anvendes i dette dokumentet, betegner en tilstrekkelig høy grad av likhet mellom to numeriske verdier (f.eks. en verdi forbundet med et antistoff ifølge oppfinnelsen og den andre forbundet med et referanse-/sammenligningsmolekyl), slik at en fagperson vil vurdere forskjellen mellom de to verdiene til å være av liten eller ingen biologisk og/eller statistisk signifikans i sammenheng med det biologiske karaktertrekket som måles med verdiene (f.eks. K d -verdier). Forskjellen mellom de to verdiene er f.eks. større enn omtrent 0 %, mindre enn omtrent 40 %, mindre enn omtrent 30 %, mindre enn omtrent 20 % og/eller mindre enn omtrent % som en funksjon av referanse-/sammenligningsverdien «Prosent (%) aminosyresekvensidentitet» og «homologi» i forbindelse med et peptid, polypeptid eller en antistoffsekvens, er definert som prosentandelen av aminosyrerester i kandidatsekvensen som er identiske med aminosyrerestene i det spesifikke peptidet eller polypeptidsekvensen, etter sammenlikning av sekvensene og innføring av åpninger hvis nødvendig, for å oppnå maksimal prosentvis sekvensidentitet, og uten å ta i betraktning eventuelle konservative substitusjoner som inngår i sekvensidentiteten. Sammenlikning for å bestemme prosent aminosyresekvensidentitet kan gjøres på forskjellige måter som er innenfor ferdighetene i faget, f.eks. ved å anvende offentlig tilgjengelig
44 dataprogramvare, som f.eks. programmene BLAST, BLAST-2, ALIGN eller MEGALIGN -(DNASTAR). Fagfolk kan bestemme egnede parametere for å måle sammenstillingen, inkludert eventuelle algoritmer som trengs for å oppnå maksimal sammenstilling gjennom hele lengden av sekvensene som sammenlignes. I forbindelse med dette dokumentet beregnes imidlertid verdier for % aminosyresekvensidentitet ved hjelp av sekvenssammenligningsprogrammet ALIGN-2, forfattet av Genentech, Inc. Kildekoden til ALIGN-2 er arkivert med brukerdokumentasjon i US Copyright Office, Washington D.C., 209, der det er registrert under U.S. Copyright-registreringsnr. TXU087. ALIGN-2-programmet er offentlig tilgjengelig gjennom Genentech, Inc., South San Francisco, California. ALIGN-2 programmet skal være utarbeidet for anvendelse på et UNIX-operativsystem, fortrinnsvis digital UNIX V4.0D. Alle sekvenssammenligningsparameterne er satt av ALIGN-2-programmet og varierer ikke. I situasjoner der ALIGN-2 benyttes til aminosyresekvenssammenligninger, beregnes % aminosyresekvensidentitet i en gitt aminosyresekvens A med eller mot en gitt aminosyresekvens B (som alternativt kan formuleres som en gitt aminosyresekvens A som har eller omfatter en viss % aminosyresekvensidentitet med eller mot en gitt aminosyresekvens B) som følger: 0 ganger fraksjonen x/y 2 30 der X er antallet aminosyrerester lagret som identiske treff av sekvenssammenligningsprogrammet ALIGN-2 under programmets sammenlikning av A og B, og der Y er det totale antallet aminosyrerester i B. Det vil være klart at hvis lengden av aminosyresekvens A ikke er lik lengden av aminosyresekvens B, vil % aminosyresekvensidentitet hos A med B ikke være lik % aminosyresekvensidentitet hos B med A. Med mindre det spesifikt er angitt noe annet fås alle verdiene for % aminosyresekvensidentitet i dette dokumentet som beskrevet i det foregående avsnittet ved hjelp av ALIGN-2-dataprogrammet. 3 Et «isolert» nukleinsyremolekyl som koder for antistoffene i dette dokumentet er et nukleinsyremolekyl som er identifisert og separert fra minst ett kontaminerende nukleinsyremolekyl som det vanligvis er forbundet med i miljøet det ble produsert i.
45 43 Fortrinnsvis er den isolerte nukleinsyren fri for assosiasjon med alle komponentene som er forbundet med produksjonsmiljøet. De isolerte nukleinsyremolekylene som koder for polypeptidene og antistoffene i dette dokumentet er i en annen form enn den formen de har eller den situasjonen de er i der de finnes i naturen. Isolerte nukleinsyremolekyler skiller seg derfor fra nukleinsyre som koder for polypeptidene og antistoffene i dette dokumentet og eksisterer naturlig i celler. Betegnelsen «kontrollsekvenser» står for DNA-sekvenser som er nødvendige for uttrykking av en operativt koblet kodingssekvens i en bestemt vertsorganisme. Kontrollsekvensene som er egnet for prokaryoter inkluderer f.eks. en promotor, eventuelt en operatørsekvens, og et ribosombindingssete. Eukaryotiske celler er kjent for å benytte promotorer, polyadenyleringssignaler og forsterkere Nukleinsyre er «operativt koblet» når den befinner seg i et funksjonelt forhold til en annen nukleinsyresekvens. F.eks. er DNA for en presekvens eller sekretorisk ledersekvens operativt koblet til DNA for et polypeptid hvis det uttrykkes som et preprotein som deltar i utskillingen av polypeptidet; en promotor eller forsterker er operativt koblet til en kodingssekvens hvis den påvirker transkriberingen av sekvensen; og et ribosombindingssete er operativt koblet til en kodingssekvens dersom det er plassert slik at det letter translateringen. Generelt betyr «operativt koblet» at DNA-sekvensene som er bundet er sammenhengende og, når det gjelder en sekretorisk leder, sammenhengende og i lesefase. Forsterkere trenger imidlertid ikke å være sammenhengende. Sammenkoblingen oppnås ved sammenbinding i hensiktsmessige restriksjonsseter. Hvis det ikke eksisterer slike seter, anvendes de syntetiske oligonukleotidadapterne eller oligonukleotidbindeleddene i samsvar med konvensjonell praksis Når betegnelsen «epitopmerket» anvendes i dette dokumentet, står den for et kimært polypeptid som omfatter et polypeptid eller antistoff beskrevet i dette dokumentet fusjonert til et «merkepolypeptid». Merkepolypeptidet har nok rester til å gi en epitop som et antistoff kan fremstilles mot, men er kort nok slik at det ikke forstyrrer aktiviteten til polypeptidet som det fuseres mot. Merkepolypeptidet er fortrinnsvis også nokså unikt, slik at antistoffet ikke i det vesentlige kryssreagerer med andre epitoper. Egnede merkepolypeptider har generelt minst seks aminosyrerester og vanligvis mellom omtrent 8 og 0 aminosyrerester (fortrinnsvis mellom omtrent og 20 aminosyrerester).
46 Slik den anvendes i dette dokumentet står betegnelsen «immunadhesin» for antistofflignende molekyler som kombinerer bindingsspesifisiteten til et heterologt protein (en «adhesjon») med effektorfunksjonene til konstante immunoglobulindomener. Strukturelt omfatter immunadhesinene en fusjon av en aminosyresekvens med den ønskede bindingsspesifisiteten som er noe annet enn antigengjenkjennelsen og bindingssetet til antistoffet (dvs. er «heterolog»), og en konstant immunoglobulindomenesekvens. Adhesindelen til et immunadhesinmolekyl er vanligvis en sammenhengende aminosyresekvens som omfatter minst bindingssetet til en reseptor eller ligand. Den konstante immunoglobulindomenesekvensen i immunadhesinet kan fås fra et hvilket som helst immunoglobulin, f.eks. IgG-1-, IgG-2-, IgG-3- eller IgG-4-undertyper, IgA (inkludert IgA-1 og IgA-2), IgE, IgD eller IgM. Ig-fusjonene inkluderer fortrinnsvis substitusjon av et domene av et polypeptid eller antistoff beskrevet i dette dokumentet i stedet for minst én variabel region i et Ig-molekyl. I en spesielt foretrukket utførelsesform inkluderer immunoglobulinfusjonen hengselet, CH2 og CH3, eller hengselet, CH1-, CH2- og CH3-regionene av et IgG1-molekyl. Se også US patent nr , utgitt 27. juni 199 når det gjelder produksjon av immunoglobulinfusjoner. I dette dokumentet inkluderer for eksempel nyttige immunadhesiner i form av sekundære legemidler som er nyttige for kombinasjonsbehandling, polypeptider som omfatter de BLySbindende delene til en BLyS-reseptor uten at transmembranen til BR3, TACI eller BCMA er fusjonert med et konstant domene av en immunoglobulinsekvens Et «fusjonsprotein» og et «fusjonspolypeptid» står for et polypeptid som har to deler som er kovalent bundet sammen, der hver av delene er et polypeptid med en egenskap som er forskjellig. Denne egenskapen kan være en biologisk egenskap, f.eks. aktivitet in vitro eller in vivo. Denne egenskapen kan også være en enkel kjemisk eller fysisk egenskap, slik som binding til et målmolekyl, katalyse av en reaksjon osv. De to delene kan være bundet direkte med en enkel peptidbinding eller være i samme leserammen ved hjelp av et peptidbindeledd. 3 En «stabil» formulering er en formulering der proteinet i den stort sett beholder sin fysiske og kjemiske stabilitet og integritet etter lagring. Forskjellige analytiske metoder for å måle proteinstabiliteten er tilgjengelige i faget og er gjennomgått i Peptide and Protein Drug Delivery, , Vincent Lee Ed., Marcel Dekker, Inc., New York, New York, pub. (1991) og Jones, A. Adv. Drug Delivery Rev. : (1993). Stabilitet kan måles ved en valgt temperatur
47 4 for en valgt tidsperiode. For hurtig screening kan formuleringen holdes ved 40 C i 2 uker til 1 måned, og ved dette tidspunktet måles stabiliteten. Hvis formuleringen skal lagres ved 2 8 C, bør formuleringen generelt være stabil ved 30 C eller 40 C i minst 1 måned og/eller stabil ved 2 8 C i minst 2 år. Hvis formuleringen skal lagres ved 30 C, bør formuleringen generelt være stabil i minst 2 år ved 30 C og/eller stabil ved 40 C i minst 6 måneder. For eksempel kan graden av aggregering under lagring anvendes som en indikator for proteinstabilitet. Dermed kan en «stabil» formulering være en formulering der mindre enn omtrent % og fortrinnsvis mindre enn omtrent % av proteinet er til stede som et aggregat i formuleringen. I andre utførelsesformer kan en eventuell økning i aggregatdannelse under lagring av formuleringen bestemmes. 1 En «rekonstituert» formulering er en formulering som har blitt fremstilt ved å løse opp et frysetørket protein eller en antistofformulering i et fortynningsmiddel, slik at proteinet dispergeres gjennom hele løsningen. Den rekonstituerte formuleringen er egnet for administrering (for eksempel parenteral administrering) til en pasient som skal behandles med proteinet av interesse, og kan være en formulering som er egnet for subkutan administrering i visse utførelsesformer ifølge oppfinnelsen En «isoton» formulering er en formulering som i det vesentlige har det samme osmotiske trykket som menneskeblod. Isotone formuleringer vil generelt ha et osmotisk trykk fra omtrent 20 til 30 mosm. Betegnelsen «hypoton» beskriver en formulering med et osmotisk trykk lavere enn menneskeblod. Tilsvarende anvendes betegnelsen «hyperton» om en formulering med høyere osmotisk trykk enn menneskeblod. Isotonisitet kan måles ved å anvende f.eks. et osmometer av damptrykk- eller isfrysingstypen. Formuleringene ifølge den foreliggende oppfinnelsen er hypertoniske fordi de er tilsatt salt og/eller buffer «Bærere» slik det anvendes i dette dokumentet, inkluderer farmasøytisk aksepterbare bærere, hjelpestoffer eller stabilisatorer som er ikke-toksiske overfor cellen eller pattedyr som eksponeres for dem ved doseringene og konsentrasjonene som benyttes. Den fysiologisk akseptable bæreren er ofte en vandig ph-bufret løsning. Eksempler på fysiologisk akseptable bærere inkluderer buffere, som f.eks. fosfat, sitrat og andre organiske syrer; antioksidanter, inkludert askorbinsyre; lett (mindre enn omtrent rester) polypeptid; proteiner, som f.eks. serumalbumin, gelatin eller immunoglobuliner; hydrofile polymerer, som f.eks. polyvinylpyrrolidon; aminosyrer, som f.eks. glycin, glutamin, asparagin, arginin eller lysin;
48 46 monosakkarider, disakkarider og andre karbohydrater, inkludert glukose, mannose eller dekstriner; kelatdannere, som f.eks. EDTA; sukkeralkoholer, som f.eks. mannitol eller sorbitol; saltdannende motioner, som f.eks. natrium; og/eller ikkeioniske surfaktanter, som f.eks. TWEEN, polyetylenglykol (PEG) og PLURONICS. En «pakningsvedlegg» står for instruksjoner som vanligvis inkluderes i kommersielle pakker av legemidler som inneholder informasjon om indikasjonene som vanligvis inkluderes i kommersielle pakker av legemidler som inneholder informasjon om indikasjonene, anvendelse, dosering, administrering, kontraindikasjoner, andre legemidler som kan kombineres med det pakkede produktet og/eller advarsler som angår anvendelsen av slike legemidler osv En «farmasøytisk akseptabel syre» inkluderer uorganiske og organiske syrer som er ikke-toksiske i konsentrasjonen og måten de formuleres med. For eksempel inkluderer egnede uorganiske syrer saltsyre, perklorsyre, hydrogenbromidsyre, hydrogenjodidsyre, salpetersyre, svovelsyre, sulfonsyre, sulfinsyre, sulfanilsyre, fosforsyre, karbonsyre osv. Egnede organiske syrer inkluderer rettkjedede og forgrenede alkyliske, aromatiske, sykliske, sykloalifatiske, arylalifatiske, heterosykliske, mettede, umettede, mono-, di- og tri-karboksylsyrer, inkludert for eksempel maursyre, eddiksyre, 2-hydroksyeddiksyre, trifluoreddiksyre, fenyleddiksyre, trimetyleddiksyre, t-butyl-eddiksyre, antranilsyre, propansyre, 2- hydroksypropansyre, 2-oksopropansyre, propandijodsyre, syklopentanpropionsyre, syklopentanpropionsyre, 3-fenylpropionsyre, butansyre, butandisyre, benzosyre, 3-(4-hydroksybenzoyl)benzosyre, 2-acetoksybenzosyre, askorbinsyre, kanelsyre, laurylsvovelsyre, stearinsyre, mukonsyre, mandelsyre, ravsyre, embonsyre, fumarsyre, eplesyre, maleinsyre, hydroksymaleinsyre, malonsyre, melkesyre, sitronsyre, vinsyre, glykolsyre, glykonsyre, glukonsyre, pyrodruesyre, glyoksalsyre, oksalsyre, metansulfonsyre, ravsyre, salisylsyre, ftalsyre, palmoinsyre, palmeinsyre, tiocyansyre, metansulfonsyre, etansulfonsyre, 1,2-etandisulfonsyre, 2-hydroksyetansulfonsyre, benzensulfonsyre, 4- klorbenzensulfonsyre, naftalen-2-sulfonsyre, p-toluensulfonsyre, kamfersulfonsyre, 4-metylbisyklo[2.2.2]-okt-2-en-1-karboksylsyre, glukoheptonsyre, 4,4'- metylenbis-3-(hydroksy-2-en-1-karboksylsyre), hydroksynaftasyre. 3 «Farmasøytisk aksepterbar syre» inkluderer uorganiske og organiske baser som er ikke-toksiske i konsentrasjonen og måten de formuleres med. For eksempel inkluderer egnede baser de som dannes av uorganiske basedannende metaller,
49 47 som f.eks. litium, natrium, kalium, magnesium, kalsium, ammonium, jern, sink, kobber, mangan, aluminium, N-metylglukamin, morfolin, piperidin og organiske ikke-toksiske baser, inkludert primære, sekundære og tertiære aminer, + substituerte aminer, sykliske aminer og basisk ionebyttermasse, [f.eks. N(R') 4 (der R' er uavhengig H eller C 1-4 -alkyl, f.eks. ammonium, Tris)], for eksempel isopropylamin, trimetylamin, dietylamin, trietylamin, tripropylamin, etanolamin, 2- dietylaminoetanol, trimetamin, disykloheksylamin, lysin, arginin, histidin, koffein, prokain, hydrabamin, kolin, betain, etylendiamin, glukosamin, metylglukamin, teobromin, puriner, piperazin, piperidin, N-etylpiperidin, polyaminpolymerer eller lignende. Spesielt foretrukne organiske ikke-toksiske baser er isopropylamin, dietylamin, etanolamin, trimetamin, disykloheksylamin, kolin og koffein. 1 Andre farmasøytisk aksepterbare syrer og baser som kan anvendes med den foreliggende oppfinnelsen inkluderer slike som er avledet fra aminosyrer, for eksempel histidin, glycin, fenylalanin, asparaginsyre, glutaminsyre, lysin og asparagin. 20 «Farmasøytisk aksepterbare» buffere og salter inkluderer slike som avledes fra både syre- og baseaddisjonssalter av de ovenfor indikerte syrene og basene. Spesifikke buffere og/eller salter inkluderer histidin, suksinat og acetat En «farmasøytisk aksepterbar sukkerart» er et molekyl som når det kombineres med et protein av interesse, i betydelig grad forhindrer eller reduserer kjemisk og/eller den fysiske ustabiliteten til proteinet ved lagring. Hvis formuleringen skal frysetørkes og deretter rekonstitueres, kan de «farmasøytisk aksepterbare sukkerartene» også omtales som en «lyoprotektant». Eksempler på sukkerarter og tilsvarende sukkeralkoholer inkluderer: en aminosyre som f.eks. mononatriumglutamat eller histidin; et metylamin som f.eks. betain; et lyotropt salt som f.eks. magnesiumsulfat; en polyol som f.eks. treverdig eller høymolekylære sukkeralkoholer, f.eks. glyserol, dekstran, erytritol, glyserol, arabitol, xylitol, sorbitol og mannitol; propylenglykol; polyetylenglykol; PLURONICS ; og kombinasjoner av disse. Andre eksempler på lyoprotektanter inkluderer glyserin og gelatin, og sukkerne melibiose, melezitose, raffinose, mannotriose og stakyose. Eksempler på reduserende sukkerarter inkluderer glukose, maltose, laktose, maltulose, isomaltulose og laktulose. Eksempler på ikke-reduserende sukkerarter inkluderer ikke-reduserende glykosider av polyhydroksyforbindelser valgt fra sukkeralkoholer og andre rettkjedede
50 48 polyalkoholer. Foretrukne sukkeralkoholer er monoglykosider, særlig forbindelsene som oppnås ved reduksjon av disakkarider som f.eks. laktose, maltose, laktulose og maltulose. Den glykosidiske sidegruppen kan være enten glukosidisk eller galaktosidisk. Andre eksempler på sukkeralkoholer er glucitol, maltitol, laktitol og isomaltulose. De foretrukne farmasøytisk aksepterbare sukkerartene er de ikke-reduserende sukkerartene trehalose eller sukrose. Farmasøytisk aksepterbare sukkerarter tilsettes til formuleringen i en «beskyttende mengde» (for eksempel forhåndsfrysetørking) som betyr at proteinet i det vesentlige beholder sin fysiske og kjemiske stabilitet og integritet under lagring (f.eks. etter rekonstitusjon og lagring) «Fortynningsmidlet» av interesse i dette dokumentet er et fortynningsmiddel som er farmasøytisk aksepterbart (sikkert og ikke-giftig for administrering til et menneske), og er nyttig for fremstilling av en flytende formulering, for eksempel en formulering som rekonstitueres etter frysetørking. Eksempler på fortynningsmidler inkluderer sterilt vann, bakteriostatisk vann for injeksjon (BWFI), en ph-bufret løsning (f.eks. fosfatbufret saltløsning), steril saltløsning, Ringers løsning eller dekstroseløsning. I en alternativ utførelsesform kan fortynningsmidler inkludere vandige løsninger av salter og/eller buffere. Et «konserveringsmiddel» er en forbindelse som kan tilsettes til formuleringene i dette dokumentet for å redusere bakterieaktiviteten. Tilsetningen av et konserveringsmiddel letter for eksempel fremstillingen av en flerbruksformulering (flerdoseformulering). Eksempler på potensielle konserveringsmidler inkluderer oktadekyldimetylbenzylammoniumklorid, heksametoniumklorid, benzalkoniumklorid (en blanding av alkylbenzyldimetylammoniumklorider der alkylgruppene er langkjedede forbindelser) og benzetoniumklorid. Andre typer konserveringsmidler inkluderer aromatiske alkoholer som f.eks. fenol, butyl og benzylalkohol, alkylparabener som for eksempel metyl- eller propylparaben, katekol, resorcinol, sykloheksanol, 3-pentanol og m-kresol. I dette dokumentet er benzylalkohol det mest foretrukne konserveringsmidlet. 3 «Behandling» står for klinisk inngripen utformet for å endre det naturlige forløpet til individet eller cellen som behandles, og kan utføres enten for profylakse eller i løpet av en klinisk patologi. Ønskelige behandlingsvirkninger inkluderer å forhindre forekomst eller tilbakefall av sykdommen, å forebygge metastaser, redusere hastigheten av sykdomsprogresjonen, bedre eller lindre
51 49 sykdomsstadiet, og tilfriskning eller forbedret prognose. I noen utførelsesformer anvendes antistoffene ifølge oppfinnelsen til å forsinke utviklingen av en sykdom eller forstyrrelse. Et individ er vellykket «behandlet», for eksempel ved å anvende de apoptotiske antistoffene mot IgE ifølge oppfinnelsen, hvis ett eller flere symptomer forbundet med en IgE-formidlet forstyrrelse er dempet En «effektiv mengde» står for minst en mengde som er effektiv, ved nødvendige doser og tidsrom, for å oppnå den ønskede eller indikerte virkningen, inkludert et terapeutisk eller profylaktisk resultat. En «terapeutisk effektiv mengde» er i det minste minimumskonsentrasjonen som kreves for å bevirke en målbar forbedring eller forhindring av en bestemt forstyrrelse. I dette dokumentet kan en terapeutisk effektiv mengde variere med faktorer som f.eks. sykdommens stadium, pasientens alder, kjønn og vekten, og antistoffets evne til å fremkalle en ønsket respons hos individet. En terapeutisk effektiv mengde er også en mengde der eventuelle toksiske eller skadelige virkninger av antistoffet mer enn oppveies av de terapeutisk fordelaktige virkningene. En «profylaktisk effektiv mengde» står for en mengde som ved nødvendige doser og tidsrom er effektiv til å oppnå det ønskede profylaktiske resultatet. Vanligvis, men ikke nødvendigvis, siden en profylaktisk dose anvendes i individer før eller på det tidligere stadiet til sykdommen, kan den profylaktisk effektive mengden være mindre enn den terapeutisk effektive mengden. 2 «Kronisk» administrering betyr administrering av legemidlet/legemidlene på en kontinuerlig i motsetning til akutt måte, for å bevare den innledende terapeutiske virkningen (aktiviteten) gjennom en lengre tidsperiode. «Periodisk» administrering er behandling som ikke gjøres fortløpende uten avbrudd, men heller er syklisk av karakter I forbindelse med behandling står «Pattedyr» for et hvilket som helst dyr som er klassifisert som et pattedyr, inkludert mennesker, husdyr og bufe, samt dyrehagedyr, sportsdyr eller kjæledyr, som hunder, hester, kaniner, kyr, griser, hamstere, ørkenrotter, mus, ildere, rotter, katter osv. Fortrinnsvis er pattedyret et menneske. Uttrykket «farmasøytisk formulering» står for et preparat som er i en slik form at den gjør det mulig for den biologiske aktiviteten til virkestoffet å være effektiv, og
52 0 som ikke inneholder noen ytterligere bestanddeler som er uakseptabelt toksiske for et individ som formuleringen skal administreres til. Slike formuleringer er sterile. En «steril» formulering er aseptisk eller fri for alle levende mikroorganismer og sporene deres Betegnelsen «omtrent» slik den anvendes i dette dokumentet, står for det vanlige feilintervallet for den angjeldende verdien som er velkjent for fagpersoner på dette tekniske området. Et «antihistamin», slik det anvendes i dette dokumentet, er et middel som motvirker den fysiologiske virkningen av histamin. Bindingen av histamin til histaminreseptorene, H 1 og H 2, fører til de karakteristiske allergiske symptomene og virkningene, eller kløe, rødhet, hevelse osv. Mange antihistaminer fungerer ved å blokkere bindingen av histamin til reseptorene sine, H1, H2, men andre antas å fungere ved å hemme frigjøring av histamin. Eksempler på antihistaminer er klorfeniramin, difenhydramin, prometazin, kromolynnatrium, astemizol, azatadinmaleat, brofeniraminmaleat, karbinoksaminmaleat, cetirizinhydroklorid, klemastinfumarat, cyproheptadinhydroklorid, deksbromfeniraminmaleat, deksklorfeniraminmaleat, dimenhydrinat, difenhydraminhydroklorid, doksylaminsuksinat, feksofenadinhydroklorid, terfenadinhydroklorid, hydroksyzinhydroklorid, loratadin, meklizinhydroklorid, tripelannaminsitrat, tripelennaminhydroklorid, triprolidinhydroklorid En «bronkodilatator» slik det anvendes i dette dokumentet, beskriver midler som motvirker eller reverserer sammentrekking av bronkiene, en fysiologisk hendelse som vanligvis forekommer i tidlig fase av astmatiske reaksjoner og fører til redusert lungekapasitet og kortpustethet. Eksempel på bronkodilatorer inkluderer epinefrin, et bredvirkende alfa- og betaadrenergt middel, og de betaadrenerge midlene albuterol, pirbuterol, metaproterenol, salmeterol og isoetarin. Bronkodilatering kan også oppnås ved administrering av xantiner, inkludert aminofyllin og teofyllin. 3 Et «glukokortikoid» slik det anvendes i dette dokumentet beskriver steroidbaserte midler som har anti-inflammatorisk aktivitet. Glukokortikoider anvendes ofte til å dempe en astmatisk reaksjon i sen fase. Eksempler på glukokortikoider inkluderer prednison, beklometasondipropionat, triamcinolonacetonid, flunisolid, betametason, budesonid, deksametason,
53 1 deksametasontramcinolon, fludrokortisonacetat, flunisolid, flutikasonpropionat, hydrokortison, prednisolon [inkludert metylprednisolon (f.eks. SOLU-MEDROL metylprednisolonnatriumsuksinat)] og triamcinolon. 1 Et «ikke-steroidisk antiinflammatorisk medikament» eller «NSAID», slik det anvendes i dette dokumentet, beskriver midler med anti-inflammatorisk aktivitet som ikke er steroidbaserte. Eksempler på NSAID inkluderer acematacin, acetaminofen, acetylsalisylsyre, azapropazon, benorylat, bromfenaknatrium, syklooksygenase(cox)-2-hemmere som GR 2303, MK966, celekoksib (CELEBREX ; 4-(-(4-metylfenyl)-3-(trifluormetyl)-1H-pyrazol-1-yl)benzensulfonamid) og valdekoksib (BEXTRA ), diklofenak, diklofenak depot, diklofenaknatrium, diflunisal, etodolak, fenbufen, fenoprofenkalsium, flurbiprofen, ibuprofen, ibuprofen depot, indometacin, ketoprofen, meklofenamatnatrium mefenaminsyre, meloksikam (MOBIC ), nabumeton, naproksen, naproksennatrium, oksyfenbutazon, fenylbutazon, piroksikam, sulindak, tenoksikam, tiaprofensyre, tolmetin, tolmetinnatrium, inkludert salter og derivater av disse osv Uttrykket «IgE-formidlede forstyrrelser» inkluderer atopiske forstyrrelser, som karakteriseres ved en generell arvelig tilbøyelighet til å reagere immunologisk på mange vanlige naturlig forekommende innåndede og svelgede antigener og kontinuerlig produksjon av IgE-antistoffer. Spesifikke atopiske forstyrrelser inkluderer allergisk astma, allergisk rhinitt (konjunktivitt), atopisk dermatitt, matallergi, anafylaksi, kontakteksem, allergisk gastroenteritt, allergisk bronkopulmonal aspergillose og allergisk purpura (Henoch-Schönlein). Atopiske pasienter har ofte flere allergier, noe som betyr at de har IgE-antistoffer mot, og symptomer fra mange miljømessige allergener, inkludert sesongallergener, helårlige og yrkesmessige allergener. Eksempel på sesongallergener inkluderer pollen (f.eks., gress, trær, rug, timotei, beiskambrosia), mens eksempler på helårlige allergener inkluderer sopp (f.eks. muggsopp, muggsoppsporer), fjær, dyr (f.eks. flass fra kjæledyr eller andre dyr) og insektrester (f.eks. støvmidd). Eksempler på yrkesmessige allergener inkluderer også dyr- (f.eks. mus) og planteantigener så vel som legemidler, vaskemidler, metaller og immunforsterkende midler som f.eks. isocyanater. Ikke-antigenspesifikke stimuli som kan føre til en IgE-formidlet reaksjon inkluderer infeksjon, irritanter som f.eks. røyk, brennbare gasser, dieseleksospartikler og svoveldioksid, mosjon, kulde og følelsesmessig stress. Spesifikke overfølsomhetsreaksjoner hos atopiske og ikke-atopiske personer med en viss genetisk bakgrunn kan skyldes eksponering for proteiner i matvarer (f.eks.
54 2 belgplanter, peanøtter), gift (f.eks. insekt-, slangegift), vaksiner, hormoner, antiserum, enzymer, lateks, antibiotika, muskelavslappende midler, vitaminer, cytotoksiner, opiater og polysakkarider som dekstrin, jerndekstran og polygelin Andre forstyrrelser som er forbundet med forhøyet IgE-konsentrasjon, som synes å være IgE-formidlede og lar seg behandle med formuleringene ifølge denne foreliggende oppfinnelsen inkluderer: ataksi-teleangiektasi, Churg-Strausssyndrom, eksem, enteritt, gastroenteritt, transplantat-mot-vert-reaksjon, hyper- IgE (Jobs) syndrom, overfølsomhet (f.eks. anafylaktisk overfølsomhet, kandidiasis, vaskulitt), IgE-myelom, inflammatorisk tarmsykdom (f.eks. Crohns sykdom, ulcerøs kolitt, ubestemmelig kolitt og smittsom kolitt), mukositt (f.eks. oral mukositt, gastrointestinal mukositt, nasal mukositt og proktitt), nekrotiserende enterokolitt og øsofagitt, parasittsykdommer (f.eks. trypanosomiase), overfølsomhetsvaskulitt, elveblest og Wiskott-Aldrich-syndrom. I tillegg inkluderer forstyrrelser som kan behandles ved å redusere IgEkonsentrasjonene, uavhengig av om forstyrrelsene i seg selv er forbundet med forhøyet IgE, og dermed bør regnes for å være innenfor omfanget av «IgE-formidlet forstyrrelse»: Addisons sykdom (kronisk binyrebarkinsuffisiens), alopesi, arvelig angioødem (Bannisters sykdom, angionevrotisk ødem), ankyloserende spondylitt, aplastisk anemi, arteritt, amyloidose, immunforstyrrelser, som f.eks. autoimmun hemolytisk anemi, autoimmun eggstokkbetennelse, autoimmun testikkelbetennelse, autoimmun polyendokrin svikt, autoimmun hemolytisk anemi, autoimmunocytopeni, autoimmun glomerulonefritt, Behcets sykdom, bronkitt, Buergers sykdom, bulløs pemfigoid, Caplans syndrom (revmatoid pneumokoniose), hjertebetennelse, cøliakisk sprue, Chediak-Higashi-syndrom, kronisk obstruktiv lungesykdom (KOLS), Cogan-Reese-syndrom (iridokornealt endotelsyndrom), CREST-syndrom, herpetiformisdermatitt (Duhrings sykdom), diabetes mellitus, eosinofil fasciitt, eosinofil nefritt, episkleritt, ekstrinsisk allergisk alveolitt, familiær paroksysmal polyserositt, Feltys syndrom, fibrosealveolitt, glomerulonefritt, Goodpastures syndrom, granulocytopeni, granulomer, granulomatose, myosittgranulomer, Graves sykdom, Guillain-Barre-syndrom (polynevritt), Hashimotos tyreoiditt (lymfadenomatøs struma), hemokromatose, histocytose, hypereosinofilt syndrom, irritabel tarm-syndrom, juvenil artritt, keratitt, spedalskhet, lupus erytematosis, Lyells sykdom, Lymes sykdom, blandet bindevevssykdom, mononevritt, mononeuritis multiplex, Muckle-Wells syndrom, mukokutant lymfeknutesyndrom (Kawasakis sykdom), multisentrisk retikulohistocystose, multippel sklerose,
55 3 myastenia gravis, mycosis fungoides, pannikulitt, pemfigoid, pemfigus, perikarditt, polynevritt, periarteritis nodosa, psoriasis, psoriasisartritt, lungeartritt, pulmonær adenomatose, lungefibrose, tilbakevendende polykondritt, giktfeber, revmatoid artritt, rhinosinusitt (bihulebetennelse), sarkoidose, skleritt, skleroserende kolangitt, serumsyke, Sézarys syndrom, Sjøgrens syndrom, Stevens-Johnsonsyndrom, systemisk mastocytose, transplantatavstøting, trombocytopenisk purpura, alymphoplasia thymica, uveitt, vitiligo, Wegeners granulomatose En «autoimmun forstyrrelse» er i dette dokumentet en sykdom eller forstyrrelse som oppstår fra og rettes mot en persons egne vev eller organer eller en kosegregering eller manifestasjon av dette eller en tilstand som skyldes dette. I mange av disse autoimmune og inflammatoriske forstyrrelsene kan det eksistere en rekke kliniske markører og laboratoriemarkører, inkludert, men ikke begrenset til, hypergammaglobulinemi, høye konsentrasjoner av autoantistoffer, antigenantistoffkompleksavsetninger i vev, utbytte av kortikosteroidbehandling eller immunundertrykkende behandlinger, og lymfoide celleaggregater i berørte vev. Uten at vi vil innskrenke oss til noen teori angående B-celleformidlet autoimmun forstyrrelse, er det antatt at B-celler viser en sykdomsfremkallende virkning ved humane autoimmune sykdommer gjennom et mangfold av mekanistiske reaksjonsveier, inkludert autoantistoffproduksjon, immunkompleksdanning, dendritt- og T-celleaktivering, cytokinsyntese, direkte kjemokinfrigjøring, og å skaffe tilholdssted for ektopisk neolymfogenese. Hver av disse reaksjonsveiene kan delta i forskjellig grad i patologien ved autoimmune sykdommer «Autoimmun sykdom» kan være en organspesifikk sykdom (dvs. at immunresponsen er spesielt rettet mot et organsystem som f.eks. det endokrine systemet, det hematopoetiske systemet, huden, det kardiopulmonale systemet, mage- og leversystemene, nyresystemet, skjoldbruskkjertelen, ørene, det nevromuskulære systemet, sentralnervesystemet osv.) eller en systemisk sykdom som kan ramme flere organsystemer (for eksempel systemisk lupus erytematosis (SLE), revmatoid artritt (RA), polymyositt osv.). Slike foretrukne sykdommer inkluderer autoimmune revmatologiske forstyrrelser (slik som f.eks. RA, Sjøgrens syndrom, skleroderma, lupus, som f.eks. SLE, og lupusnefritt, polymyosittdermatomyositt, kryoglobulinemi, fosfolipidantistoffsyndrom og psoriasisartritt), autoimmune mage- og leverforstyrrelser (som for eksempel inflammatoriske tarmsykdommer (f.eks. ulcerøs kolitt og Crohns sykdom),
56 autoimmun gastritt og pernisiøs anemi, autoimmun hepatitt, primær biliær cirrhose, primær skleroserende kolangitt og cøliaki), vaskulitt (slik som f.eks. ANCA-negativ vaskulitt og ANCA-forbundet vaskulitt, inkludert Churg-Strauss vaskulitt, Wegeners granulomatose og mikroskopisk polyangitt), autoimmune nevrologiske forstyrrelser (som for eksempel multippel sklerose, opsoklonusmyoklonussyndrom, myasthenia gravis, Devics sykdom, Parkinsons sykdom, Alzheimers sykdom og autoimmune polynevropatier), nyreforstyrrelser (som for eksempel glomerulonefritt, Goodpastures syndrom og Bergers sykdom), autoimmune dermatologiske forstyrrelser (slik som for eksempel psoriasis, elveblest, pemfigus vulgaris, bulløs pemfigoid og kutan lupuserytematose), hematologiske forstyrrelser (som for eksempel trombocytopenisk purpura, trombotisk trombocytopenisk purpura, purpura etter transfusjon og autoimmun hemolytisk anemi), aterosklerose, uveitt, autoimmune hørselssykdommer (som for eksempel sykdommer i det indre øret og hørselstap), Behcets sykdom, Raynauds syndrom, organtransplantasjon og autoimmune endokrine forstyrrelser (som for eksempel diabetesrelaterte autoimmune sykdommer som f.eks. insulinavhengig diabetes mellitus (IDDM), Addisons sykdom og autoimmun skjoldkjertelsykdom (f.eks. Graves' sykdom og tyreoiditt)). Mer foretrukne slike sykdommer inkluderer for eksempel RA, ulcerøs kolitt, ANCAforbundet vaskulitt, lupus, multippel sklerose, Sjøgrens syndrom, Graves' sykdom, IDDM, pernisiøs anemi, tyreoiditt og glomerulonefritt. 2 Uttrykket «cytotoksisk middel» slik det anvendes i dette dokumentet, står for et stoff som hemmer eller hindrer funksjonen til celler og/eller forårsaker ødeleggelse av celler. Uttrykket inkluderer radioaktive isotoper (f.eks. At 211, I 131, I 12, Y 90, Re 186, Re 188, Sm 13, Bi 212, P 32 og radioaktive isotoper av Lu), og toksiner som f.eks. småmolekylære toksiner eller enzymatisk aktive toksiner fra bakterier, sopp, planter eller dyr, eller fragmenter av disse Et «kjemoterapeutikum» er en kjemisk forbindelse som kan anvendes til å behandle kreft. Eksempler på kjemoterapeutiske midler inkluderer alkylerende midler som f.eks. tiotepa og syklofosfamid (CYTOXAN ); alkylsulfonater som f.eks. busulfan, improsulfan og piposulfan; aziridiner som f.eks. benzodopa, carboquone, meturedopa og uredopa; etyleniminer og metylmelaminer, inkludert altretamin, trietylenmelamin, trietylenfosforamid, trietylentiofosforamid og trimetylolomelamin; acetogeniner (særlig bullatacin og bullatacinon); delta-9-tetrahydrokannabinol (dronabinol, MARINOL ); beta-
57 lapakon; lapakol; kolkisiner; betulinsyre; et kamptotecin (inkludert den syntetiske analogene topotekan (HYCAMTIN ), CPT-11 (irinotekan, CAMPTOSAR ), acetylkamptotecin, skopolektin og 9-aminokamptotecin); bryostatin; pemetreksed; callystatin; CC-6 (inkludert de syntetiske adozelesin-, carzelesin- og bizelesinanalogene); podofyllotoksin; podofyllinsyre; teniposid; kryptofyciner (særlig kryptofycin 1 og kryptofycin 8); dolastatin; duokarmycin (inkludert de syntetiske analogene KW-2189 og CB1- TM1); eleuterobin; pankratistatin; TLK-286; CDP323, en oral alfa-4- integrinhemmer; et sarkodiktyin; spongistatin; nitrogensennep, f.eks. klorambucil, klomafazin, kolofosfamid, estramustin, ifosfamid, mekloretamin, mekloretaminoksidhydroklorid, melfalan, novembikin, fenesterin, prednimustin, trofosfamid, uracilsennep; nitrosoureaforbindelser som f.eks. karmustin, klorzotocin, fotemustin, lomustin, nimustin og ranimnustin; antibiotika som f.eks. endiynantibiotika (f.eks. calicheamicin, spesielt calicheamicin gammaii og calicheamicin omegai1 (se f.eks. Nicolaou et al., Angew. Chem Intl. Ed. Engl., 33: (1994)); dynemicin, inkludert dynemicin A; et esperamicin; så vel som neokarzinostatinkromofor og beslektede antibiotiske kromoproteinendiynkromoforer), aklacinomyciner, aktinomycin, autramycin, azaserin, bleomyciner, kaktinomycin, karabicin, karminomycin, karzinofilin, kromomyciner, daktinomycin, daunorubicin, detorubicin, 6-diazo--okso-Lnorleucin, doksorubicin (inkludert ADRIAMYCIN, morfolinodoksorubicin, cyanomorfolinodoksorubicin, 2-pyrrolinodoksorubicin, doksorubicin-hciliposominjeksjon (DOXIL ) og deoksydoksorubicin), epirubicin, esorubicin, idarubicin, marcellomycin, mitomyciner som f.eks. mitomycin C, mykofenolsyre, nogalamycin, olivomyciner, peplomycin, potfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin; antimetabolitter som f.eks. metotreksat, gemcitabin (GEMZAR ), tegafur (UFTORAL ), kapecitabin (XELODA ), et epotilon og -fluoruracil (-FU); folsyreanaloger som f.eks. denopterin, metotreksat, pteropterin, trimetreksat; purinanaloger som f.eks. fludarabin, 6- merkaptopurin, tiamiprin, tioguanin; pyrimidinanaloger som f.eks. et ancitabin, azacitidin, 6-azauridin, karmofur, cytarabin, dideoksyuridin, doksifluridin, enocitabin, floksuridin og imatinib (et 2-fenylaminopyrimidinderivat), så vel som andre c-kit-hemmere; antiadrenergika som f.eks. aminoglutetimid, mitotan, trilostan; folsyretilskudd som f.eks. frolinsyre; aceglaton; aldofosfamidglykosid; aminolevulinsyre; eniluracil; amsakrin; bestrabucil; bisantren; edatraksat; defofamin; demekolcin; diazikvon; elfornitin;
58 elliptiniumacetat; etoglucid; galliumnitrat; hydroksyurea; lentinan; lonidainin; maytansinoider som f.eks. maytansin og ansamitociner; mitoguazon; mitoksantron; mopidanmol; nitraerin; pentostatin; fenamet; pirarubicin; losoksantron; 2-etylhydrazid; prokarbazin; PSK -polysakkaridkompleks (JHS Natural Products, Eugene, Oregon); razoksan; rhizoksin; sizofiran; spirogermanium; tenuazonsyre; triazikvon; 2,2',2"-triklortrietylamin; trikotecener (særlig T-2-toksin, verracurin A, roridin A og anguidin), uretan; vindesin (ELDISINE, FILDESIN ); dakarbazin, mannomustin, mitobronitol, mitolaktol, pipobroman; gacytosin; arabinosid («Ara-C»); tiotepa; taksoider, f.eks. paklitaxel (TAXOL ), albuminutviklet nanopartikkelformulering av paklitaxel (ABRAXANE ) og docetaxel (TAXOTERE ); klorambucil; 6- tioguanin; merkaptopurin; metotreksat; platinaanaloger som f.eks. cisplatin og karboplatin, vinblastin (VELBAN ); platina; etoposid (VP-16); ifosfamid; mitoksantron; vinkristin (ONCOVIN ); oksaliplatin; leukovovin; vinorelbin (NAVELBINE ), novantron; edatreksat; daunomycin; aminopterin; ibandronat; topoisomerasehemmer RFS 2000; difluormetylornitin (DMFO); retinoider, som f.eks. retinsyre, farmasøytisk aksepterbare salter, syrer eller derivater av en hvilken som helst av de ovennevnte, så vel som kombinasjoner av to eller flere av de ovennevnte, som f.eks. CHOP, en forkortelse for en kombinert behandling med syklofosfamid, doksorubicin, vinkristin og prednisolon, og FOLFOX, en forkortelse for et behandlingsregime med oksaliplatin (ELOXATIN ) i kombinasjon med -FU og leukovovin Også inkludert i denne definisjonen er antihormonelle midler som virker ved å regulere, redusere, blokkere eller hemme virkningene av hormonene som kan fremme veksten av kreft, og er ofte i form av systemisk eller helkroppsbehandling. De kan være hormoner i seg selv. Eksempler inkluderer antiøstrogener og selektive østrogenreseptormodulatorer (SERM-er), inkludert for eksempel tamoksifen (inkludert NOLVADEX -tamoksifen), raloksifen (EVISTA ), droloksifen, 4-hydroksytamoksifen, trioksifen, keoksifen, LY117018, onapriston og toremifen (FARESTON ); antiprogesteroner; nedregulatorer for østrogenreseptorer (ERD); østrogenreseptorantagonister som f.eks. fulvestrant (FASLODEX ); midler som virker ved å undertrykke eller «slå av» eggstokkene, for eksempel agonister for det luteiniserende hormon-frigjørende hormonet (LHRH) som f.eks. leuprolidacetat (LUPRON og ELIGARD ), goserelinacetat, buserelinacetat og tripterelin; antiandrogener som f.eks. flutamid, nilutamid og bikalutamid; og aromatasehemmere som hemmer enzymet aromatase som
59 regulerer østrogenproduksjonen i binyrekjertlene, f.eks. 4()-imidazoler, aminoglutetimid, megestrolacetat (MEGASE ), eksemestan (AROMASIN ), formestan, fadrozol, vorozol (RIVISOR ), letrozol (FEMARA ) og anastrozol (ARIMIDEX ). I tillegg inkluderer en slik definisjon av kjemoterapeutiske midler bisfosfonater, som f.eks. klodronat (for eksempel BONEFOS eller OSTAC ), etidronat (DIDROCAL ), NE-809, zoledronsyre/zoledronat (ZOMETA ), alendronat (FOSAMAX ), pamidronat (AREDIA ), tiludronat (SKELID ) eller risedronat (ACTONEL ); så vel som troksacitabin (en cytosinanalog til 1,3- dioksolannukleosid); antisense-oligonukleotider, særlig slike som hemmer uttrykking av gener i signalreaksjonsveier som er involvert i avvikende celleproliferering, som for eksempel PKC-alfa, Raf, H-Ras, og reseptoren for den epidermale vekstfaktoren (EGF-R); vaksiner som f.eks. THERATOPE -vaksine og genterapivaksiner, for eksempel ALLOVECTIN -vaksine, LEUVECTIN -vaksine og VAXID -vaksine; topoisomerase-1-hemmer (f.eks. LURTOTECAN ); et antiøstrogen som f.eks. fulvestrant; en KIT-hemmer som f.eks. imatinib eller EXEL-0862 (en tyrosinkinasehemmer); EGFR-hemmer som f.eks. erlotinib eller cetuksimab; en anti-vegf-hemmer som f.eks. bevacizumab; arinotekan; rmrh (f.eks. ABARELIX ); lapatinib og lapatinibditosylat (en småmolekylær hemmer av dobbeltvirkende ErbB-2- og EGFR-tyrosinkinaser som også er kjent som GW72016); 17AAG (geldanamycinderivat som er et giftstoff mot varmesjokkprotein (HSP) 90) og farmasøytisk aksepterbare salter, syrer eller derivater av hvilke som helst av de ovennevnte Et «veksthemmende middel» står for en forbindelse eller sammensetning som hemmer en cellevekst som er avhengig av reseptoraktivering, enten in vitro eller in vivo. Dermed inkluderer det veksthemmende midlet et middel som i vesentlig grad reduserer prosentandelen av reseptoravhengige celler i S-fasen. Eksempler på veksthemmende midler inkluderer midler som blokkerer cellesyklusprogresjonen (på et annet sted enn S-fasen), f.eks. midler som utløser G1-stans og M-fasestans. Klassiske M-faseblokkere inkluderer vincaene og vinkaalkaloidene (vinkristin og vinblastin), taxaner og topoisomerase-ii-hemmere som f.eks. doksorubicin, epirubicin, daunorubicin, etoposid og bleomycin. Midlene som stanser G1 «renner» også over på S-fasestansen, for eksempel DNAalkylerende midler som f.eks. tamoksifen, prednison, dakarbazin, mekloretamin, cisplatin, metotreksat, -fluoruracil og ara-c. Flere opplysninger kan finnes i The Molecule Basis of Cancer, Mendelsohn and Israel, red., kapittel 1, kalt «Cell cycle regulation, oncogenes, and antineoplastic drugs», av Murakami et al. (WB
60 8 1 Saunders: Philadelphia, 199), særlig s. 13. Taxanene (paklitaxel og docetaxel) er legemidler mot kreft, begge avledet fra barlindtreet. Docetaksel (TAXOTERE, Rhone-Poulenc Rorer), avledet fra den europeiske barlinden, er en halvsyntetisk analog av paklitaxel (TAXOL, Bristol-Myers Squibb). Betegnelsen «cytokin» er en fellesbetegnelse for proteiner frigjort av en cellepopulasjon som opptrer på en annen celle som intercellulære formidlere. Eksempler på slike cytokiner er lymfokiner, monokiner; interleukiner (IL) som f.eks. IL-1, IL-1α, IL-2, IL-3, IL-4, IL-, IL-6, IL-7, IL-8, IL-9, IL-11, IL-12, IL-13, IL-1, inkludert PROLEUKIN ril-2; en tumornekrosefaktor som f.eks. TNF-α eller TNF-β; og andre polypeptidfaktorer, inkludert LIF- og KIT-liganden (KL). Slik den anvendes i dette dokumentet inkluderer betegnelsen cytokin proteiner fra naturlige kilder eller fra rekombinant cellekultur og biologisk aktive ekvivalenter av de opprinnelige sekvenscytokinene, inkludert syntetisk produserte småmolekylære stoffer og farmasøytisk aksepterbare derivater og salter av disse Betegnelsen «hormon» står for polypeptidhormoner, som generelt skilles ut av kjertelorganer med kanaler. Blant hormonene inkluderes for eksempel veksthormon, som humant veksthormon, N-metionyl-humant veksthormon, og bovint veksthormon; paratyreoideahormon; tyroksin; insulin; proinsulin; relaksin; østradiol; hormonerstatningsterapi; androgener som f.eks. kalusteron, dromostanolonpropionat, epitiostanol, mepitiostan eller testolakton; prorelaksin; glykoproteinhormoner som f.eks. follikkelstimulerende hormon (FSH), tyreoideastimulerende hormon (TSH), og luteiniserende hormon (LH); prolaktin, placentalt laktogen, murint gonadotropinforbundet peptid, gonadotropinfrigjørende hormon; inhibin; aktivin; müllerskhemmende stoff; og trombopoetin. Slik den anvendes i dette dokumentet inkluderer hormonbetegnelsen proteiner fra naturlige kilder eller fra rekombinant cellekultur og biologisk aktive ekvivalenter av hormonet med den opprinnelige sekvensen, inkludert syntetisk produserte småmolekylære stoffer og farmasøytisk aksepterbare derivater og salter av disse. 3 Betegnelsen «vekstfaktor» står for proteiner som fremmer vekst, og inkluderer for eksempel levervekstfaktor; fibroblastvekstfaktor; vaskulær endotelvekstfaktor; nervevekstfaktorer, som f.eks. NGF-β; blodplateavledet vekstfaktor; transformerende vekstfaktorer (TGF) som f.eks. TGF-α og TGF-β; insulinlignende vekstfaktor I og II; erytropoetin (EPO); osteoinduktive faktorer; interferoner, som f.eks. interferon α, β og γ; og kolonistimulerende faktorer
61 9 (CSF) som f.eks. makrofag-csf (M-CSF); granulocyttmakrofag-csf (GM-CSF); og granulocytt-csf (G-CSF). Slik den anvendes i dette dokumentet inkluderer vekstfaktorbetegnelsen proteiner fra naturlige kilder eller fra rekombinant cellekultur og biologisk aktive ekvivalenter av vekstfaktoren med den opprinnelige sekvensen, inkludert syntetisk produserte småmolekylære stoffer og farmasøytisk aksepterbare derivater og salter av disse Betegnelsen «integrin» står for et reseptorprotein som lar celler både binde seg til og respondere på den ekstracellulære matriksen, og som er involvert i en rekke cellefunksjoner som sårheling, celledifferensiering, spesifikk vevssøking hos tumorceller og apoptose. De er en del av en stor familie av celleadhesjonsreseptorer som er involvert i interaksjoner mellom cellen og den ekstracellulære matriksen og mellom celler. Funksjonelle integriner består av to transmembrane glykoproteinunderenheter, kalt alfa og beta, som er ikke-kovalent bundet. Alfaunderenhetene har alle en viss homologi med hverandre, liksom betaunderenhetene. Reseptorene inneholder alltid en alfakjede og en betakjede. Eksempler inkluderer Alpha6beta1, Alpha3beta1, Alpha7beta1, LFA-1 osv. Slik den anvendes i dette dokumentet inkluderer «integrin»-betegnelsen proteiner fra naturlige kilder eller fra rekombinant cellekultur og biologisk aktive ekvivalenter av integrinet med den opprinnelige sekvensen, inkludert syntetisk produserte småmolekylære stoffer og farmasøytisk aksepterbare derivater og salter av disse En «TNF-antagonist» defineres i dette dokumentet som et molekyl som reduserer, blokkerer, hemmer, opphever eller forstyrrer TNFa-aktiviteten in vitro, in situ, og/eller fortrinnsvis in vivo. En egnet TNF-antagonist kan også redusere, blokkere, oppheve, forstyrre, forhindre og/eller hemme TNF RNA, DNA eller proteinsyntesen, TNFa-frigjøringen, TNFa-reseptorsignaleringen, membranenes TNFα-spalting, TNFα-aktiviteten, TNFα-produksjonen og/eller syntesen. Slike TNF-antagonister inkluderer, men er ikke begrenset til, antistoffer mot TNFα, antigenbindende fragmenter av disse, spesifiserte mutanter eller domener av disse som spesifikt binder TNFα, og som ved binding til TNFα ødelegger eller desimerer celler som uttrykker TNFα i et pattedyr, og/eller forstyrrer én eller flere av funksjonene til disse cellene; en løselig TNF-reseptor (for eksempel p, p70 eller P8) eller fragment av denne, fusjonspolypeptider, en TNF-antagonist for et lite molekyl, for eksempel TNFbindende protein I eller II (TBP-I eller TBP-II), nerelimonmab, CDP-71, infliksimab, enteracept (ENBREL ), adalimulab (HUMIRA ), CDP-71, CDP-
62 60 870, afelimomab, lenercept eller lignende), antigenbindende fragmenter av disse, og reseptormolekyler som binder spesifikt til TNFα; forbindelser som forhindrer og/eller hemmer TNFα-syntese, TNFα-frigjøring eller virkningen av den på målceller, for eksempel talidomid, tenidap, fosfodiesterasehemmere (f.eks. pentoksifyllin og rolipram), A2b-adenosinreseptoragonister og A2badenosinreseptorforsterkere; forbindelser som hindrer og/eller hemmer TNFαreseptorsignalisering, for eksempel kinsasehemmere av mitogenaktivert protein (MAP); forbindelser som blokkerer og/eller hemmer spalting av membran-tnfα, for eksempel metalloproteinasehemmere; forbindelser som blokkerer og/eller hemmer TNFα-aktiviteten, for eksempel hemmere av angiotensinomdannende enzym (ACE) (f.eks. kaptopril); og forbindelser som blokkerer og/eller hemmer TNFα-produksjon og/eller syntese, for eksempel MAP-kinasehemmere. Den foretrukne antagonisten omfatter et antistoff «Tumornekrosefaktor-alfa», «TNF-a» eller «TNFα» står for et humant TNFαmolekyl som omfatter aminosyresekvensen til Pennica et al., Nature, 312:721 (1984) eller Aggarwal et al., JBC, 260:234 (198). I dette dokumentet er en «TNFα-hemmer» et middel som, til en viss grad, hemmer en biologisk funksjon hos TNFα, generelt ved å bindes til TNFα og nøytralisere aktiviteten dens. I dette dokumentet inkluderer eksempler på TNFα-hemmere etanercept (ENBREL ), infliksimab (REMICADE ) og adalimumab (HUMIRA ) I dette dokumentet inkluderer eksempler på «integrinantagonister eller - antistoffer» et LFA-1-antistoff, som f.eks. efalizumab (RAPTIVA ), kommersielt tilgjengelig fra Genentech, eller et alfa-4-integrinantistoff som f.eks. natalizumab (ANTEGREN ) tilgjengelig fra Biogen, eller diazasykliske fenylalaninderivater (WO 2003/894), fenylalaninderivater (WO 2003/70709, WO 2002/28830, WO 2002/16329 and WO 2003/3926), fenylpropionsyrederivater (WO 2003/13), enaminderivater (WO 2001/79173), propansyrederivater (WO 2000/37444), alkansyrederivativer (WO 2000/327), substituerte fenylderivater (US pat. nr og ), aromatiske aminderivater (US pat. nr ), ADAMdisintegrindomenepolypeptider (US2002/ ), antistoffer mot alfa-v-beta3- integrin (EP 63394), azabrodannede bisykliske aminosyrederivater (WO 2002/026) osv. Uttrykket «immunundertrykkende middel» står i dette dokumentet for et stoff som virker ved å undertrykke eller maskere immunsystemet til individet som behandles.
63 Dette vil inkludere stoffer som undertrykker cytokinproduksjonen, nedregulerer eller undertrykker selv-antigenuttrykkingen eller maskerer MHC-antigener. Eksempler på slike midler inkluderer 2-amino-6-aryl--substituerte pyrimidiner (se U.S ); ikke-steroidiske antiinflammatoriske legemidler (NSAID-er); ganciklovir, takrolimus, glukokortikoider, som f.eks. kortisol eller aldosteron, antiinflammatoriske midler som f.eks. en syklooksygenasehemmer, en - lipoksygenasehemmer eller en leukotrienreseptorantagonist; purinantagonister som f.eks. azatioprin eller mykofenolatmofetil (MMF); trokade (Ro32-3); en perifer sigmareseptorantagonist som f.eks. ISR-31747; alkylerende midler som f.eks. syklofosfamid; bromkryptin; danazol; dapson; glutaraldehyd (som maskerer MHCantigenene, som beskrevet i U.S ); anti-idiotypiske antistoffer for MHCantigener og MHC-fragmenter; syklosporin A; steroider som f.eks. kortikosteroider eller kortikosteroider eller glukokortikoidanaloger, for eksempel prednison, metylprednisolon, inkludert SOLU-MEDROL -metylprednisolonnatriumsuksinat, rimeksolon og deksametason; dihydrofolatreduktasehemmere som f.eks. metotreksat (oralt eller subkutant); legemidler mot malaria som f.eks. klorokin og hydroksyklorokin; sulfasalazin; leflunomid; cytokinfrigjøringshemmere som f.eks. SB-2396 og SB monoklonale antistoffer og en MHC II-antagonist som f.eks. ZD231; en PG1-reseptorantagonist som f.eks. ZD493; en VLA4- adhesjonsblokkerer som ZD7349; antistoffer mot cytokin eller cytokinreseptorer, inkludert antistoffer mot interferon alfa, beta eller gamma, anti-tnf-α-antistoffer (infliksimab (REMICADE ) eller adalimumab), antistoffer mot TNF-α-immunadhesin (etanercept), mot TNF-beta, blokkere av interleukin-1 (IL-1) som f.eks. rekombinant HuIL-1Ra- og IL-1B-hemmer, antistoffer mot interleukin-2 (IL-2) og IL-2-reseptoren; IL-2-fusjonstoksin; antistoffer mot L3T4; leflunomid; heterologt antilymfocyttglobulin; OPC-1497; NISV (immunresponsmodifikatorer); en essensiell fettsyre som f.eks. gammalinolensyre eller ikosapentaensyre; CD-4- blokkere, pan-t-antistoffer, fortrinnsvis antistoffer mot CD3 eller CD4/CD4a; kostimulerende modifikatorer (f.eks. CTLA4-Fc-fusjon, også kjent som ABATACEPT ; antistoffer og antagonister mot interleukin-6(il-6)-reseptoren, antistoffer mot LFA-1, inkludert antistoffer mot CD11a og CD18; løselig peptid som inneholder et LFA-3-bindende domene (WO 1990/08187); streptokinase; IL-; antagonister mot IL-4, antagonister mot IL-13 og bispesifikke antagonistantistoffer mot IL-4/IL-13, transformerende vekstfaktor beta (TGF-beta); streptodornase; RNA eller DNA fra verten; FK06; RS-61443; enlimomab; CDP-8; PNP-hemmer; CH- 3298; GW33430; 4162W94, klorambucil; deoksyspergualin; rapamycin; T- cellereseptor (US ); T-cellereseptorfragmenter (Offner et al., Science,
64 62 21: (1991); WO 1990/11294; Janeway, Nature, 341: (1989); og WO 1991/01133); BAFF-antagonister, som f.eks. BAFF-antistoffer og BR3- antistoffer; ztnf4-antagonister (Mackay and Mackay, Trends Immunol., 23:113- (2002)); biologiske midler som forstyrrer T-cellehjelpersignalene, som f.eks. anti- CD40-reseptor eller anti-cd40-ligand (CD 14), inkludert blokkerende antistoffer til CD40-CD40-liganden (f.eks. Durie et al., Science, 261: (1993); Mohan et al., J. Immunol., 14: (199)) og CTLA4-Ig (Finck et al., Science, 26: (1994)); og T-cellereseptorantistoffer (EP 340,9), som f.eks. TB9. Noen foretrukne immunundertrykkende midler i dette dokumentet inkluderer syklofosfamid, klorambucil, azatioprin, leflunomid, MMF eller metotreksat (MTX). 1 «Sykdomsmodifiserende antirevmatiske legemidler» eller «DAMARD» inkluderer for eksempel klorokin, hydroksyklorokin, myokrisin, auranofin, sulfasalazin, metotreksat, leflunomid, etanercept, infliksimab (og oralt og subkutant MTX), azatioprin, D-penicilamin, gullsalter (orale), gullsalter (intramuskulære), minosyklin, syklosporin, f.eks. syklosporin A og topisk syklosporin, stafylokokkprotein A Goodyear and Silverman, J. Exp. Med., 197: (2003)), inkludert salter og derivater av disse osv. 20 En «B-celle» er en lymfocytt som modnes i beinmargen, og inkluderer en naiv B-celle, hukommelses-b-celle eller effektor-b-celle (plasmaceller). B-cellen i dette dokumentet kan være normal eller ikke-ondartet En «B-celleoverflatemarkør» og et «B-celleoverflateantigen» er i dette dokumentet et antigen uttrykt på overflaten av en B-celle som kan målrettes med en antagonist som binder seg til den. Eksempler på B- celleoverflatemarkører inkluderer leukocyttoverflatemarkørene CD, CD19, CD20, CD21, CD22, CD23, CD24, CD37, CD40, CD3, CD72, CD73, CD74, CDw7, CDw76, CD77, CDw78, CD79a, CD79b, CD80, CD81, CD82, CD83, CDw84, CD8 og CD86 (beskrivelser finnes i The Leukocyte Antigen Facts Book, 2. utgave. 1997, utg. Barclay et al. Academic Press, Harcourt Brace & Co., New York). Andre B-celleoverflatemarkører inkluderer RP, FcRH2, B- cell CR2, CCR6, P2X, HLA-DOB, CXCR, FCER2, BR3, Btig, NAG14, SLGC16270, FcRH1, IRTA2, ATWD78, FcRH3, IRTA1, FcRH6, BCMA og Den foretrukne B-celleoverflatemarkøren uttrykkes heller på B-celler enn på andre ikke-b-cellevev i et pattedyr, og kan uttrykkes på både forløperog modne B-celler. De mest foretrukne av disse markørene er CD20 og CD22.
65 63 «CD20»-antigenet, eller «CD20», er et ikke-glykosylert fosfoprotein på rundt 3 kda som finnes på overflaten av over 90 % av B-cellene fra perifert blod eller lymfeorganer. CD20 er til stede både på normale B-celler og ondartede B-celler, men uttrykkes ikke på stamceller. Andre navn på CD20 i litteraturen inkluderer «B-lymfocyttbegrenset antigen» og «Bp3». CD20-antigenet er beskrevet for eksempel i Clark et al., Proc. Natl. Acad. Sci. (USA) 82:1766 (198). 1 «CD22»-antigenet eller «CD22», også kjent som BL-CAM eller Lyb8, er et innebygd membranglykoprotein av type 1 med molekylvekt på rundt 130 (redusert) til 140 kd (ikke-redusert). Det uttrykkes i både cytoplasmaet og cellemembranen til B-lymfocytter. CD22-antigenet fremkommer tidlig i B- cellelymfocyttdifferensieringen, omtrent på samme stadiet som CD19-antigenet. I motsetning til andre B-cellemarkører begrenses CD22-membranuttrykkingen til de sene differensieringsstadiene som omfattes mellom modne B-celler (CD22+) og plasmaceller (CD22 ). CD20-antigenet er beskrevet f.eks. i Wilson et al., J. Exp. Med. 173:137 (1991) og Wilson et al., J. Immunol. :013 (1993) Et «antistoff som binder til en B-celleoverflatemarkør» er et molekyl som, ved å binde seg til en B-celleoverflatemarkør, ødelegger eller desimerer B-celler i et pattedyr og/eller forstyrrer én eller flere B-cellefunksjoner, f.eks. ved å redusere eller forhindre en humoral respons utløst av B-cellen. Antistoffet er fortrinnsvis i stand til å desimere B-celler (dvs. redusere sirkulerende B-cellekonsentrasjoner) i et pattedyr som behandles med det. Slik desimering kan oppnås ved forskjellige mekanismer, f.eks. antistoffavhengig celleformidlet cytotoksisitet (ADCC) og/eller komplementavhengig cytotoksisitet (CDC), hemming av B- celleproliferasjonen og/eller utløsing av B-celledød (f.eks. ved apoptose) Eksempler på CD20-antistoffer inkluderer: «C2B8», som nå heter «rituximab» («RITUXAN») (US patent nr ); det yttrium-[90]-merkede murine 2B8-antistoffet som betegnes «Y2B8» eller «ibritumomab tiuxetan» (ZEVALIN ) kommersielt tilgjengelig fra IDEC Pharmaceuticals, Inc. (US patent nr ; 2B8 deponert hos ATCC under tilvekstnr. HB11388 den 22. juni 1993), murint IgG2a «B1», også kalt «tositumomab», eventuelt merket med 131I for å lage «131I-B1»- eller «jod-i131-tositumomab»- antistoffet (BEXXAR ) kommersielt tilgjengelig fra Corixa (se også US patent nr ); murint monoklonalt antistoff «1F» (Press et al. Blood
66 (2):84-91 (1987) og varianter av dette inkludert «rammelappet» eller humanisert 1F (WO 2003/002607, Leung, S.; ATCC-deponering HB-9640); murint 2H7- og kimært 2H7-antistoff (US patent nr ); et humanisert 2H7 (WO 2004/06312 (Lowman et al.) og som utlagt nedenfor); HUMAX- CD20, fullt humant, høyaffinitetsantistoff målrettet mot CD20-molekylet i cellemembranen til B-cellene (Genmab, Danmark; se f.eks. Glennie and van de Winkel, Drug Discovery Today 8: 03 (2003) og Cragg et al., Blood 1: 4 2 (2003)); de humane monoklonale antistoffene er utlagt i WO04/03607 (Teeling et al.); AME-133 -antistoffer (Applied Molecular Evolution); A20-antistoff eller varianter av dette, som f.eks. kimært eller humanisert A20-anistoff (henholdsvisca20, ha20) (US 2003/ , Immunomedics); og de monoklonale antistoffene L27, G28-2, 93-1B3, B-C1 eller NU-B2 tilgjengelig fra the International Leukocyte Typing Workshop (Valentine et al., i: Leukocyte Typing III (McMichael, red., s. 440, Oxford University Press (1987)). De foretrukne CD20-antistoffene i dette dokumentet er kimære, humaniserte eller humane CD20-antistoffer, og mer foretrukne er rituximab, et humanisert 2H7, kimært eller humanisert A20-antistoff (Immunomedics) og HUMAX-CD20 -humant CD20-antistoff (Genmab). 20 Betegnelsene «rituximab» eller «RITUXAN» står i dette dokumentet for til det genetisk konstruerte kimære murint/humane monoklonale antistoffet som er rettet mot CD20-antigenet og betegnes «C2B8» i US patent nr , inkludert fragmenter av disse som beholder evnen til å binde CD Bare her i dette dokumentet og med mindre det er angitt noe annet, står et «humanisert 2H7» for et humanisert CD20-antistoff, eller et antigenbindende fragment av dette der antistoffet er effektivt til å desimere primat-b-celler in vivo. Antistoffet inkluderer slike som er utlagt i US 2006/ og figurene der, og inkludert versjon 114, med sekvenser som tilveiebringes i US 2006/ Se også US 2006/ og US 2006/ I en oppsummering av forskjellige foretrukne utførelsesformer ifølge oppfinnelsen vil V-regionen av variantene basert på 2H7-versjon 16 som er offentliggjort i US 2006/ ha aminosyresekvensene til v16 bortsett fra ved de posisjonene for aminosyresubstitusjoner som er angitt i tabellen nedenfor. Med mindre det er angitt noe annet, vil 2H7-variantene ar den samme L-kjeden som v16.
67 6 2H7- Endringer i den tunge Endringer i den lette Fc-endringer versjon kjeden (V H ) kjeden (V L ) S298A, E333A, K334A 73 N0A M32L 7 N0A M32L S298A, E333A, K334A 96 D6A, N0A S92A 114 D6A, N0A M32L, S92A S298A, E333A, K334A 11 D6A, N0A M32L, S92A S298A, E333A, K334A, E36D, M38L 116 D6A, N0A M32L, S92A S298A, K334A, K322A 138 D6A, N0A M32L, S92A S298A, E333A, K334A, K326A 477 D6A, N0A M32L, S92A S298A, E333A, K334A, K326A, N434W K334L Et foretrukket humanisert 2H7 er et intakt antistoff eller antistoffragment som har sekvensen til versjon 16. Et annet foretrukket humanisert 2H7 har sekvensene til versjon 114. «BAFF-antagonister» er et hvilket som helst molekyl som blokkerer aktiviteten til BAFF eller BR3. De inkluderer immunadhesiner som omfatter en del av BR3, TACI eller BCMA som binder BAFF eller varianter av disse, som binder BAFF. I andre aspekter er BAFF-antagonisten et BAFF-antistoff. Et «BAFF-antistoff» er et antistoff som binder BAFF, og fortrinnsvis binder BAFF innenfor en region av humant BAFF som omfatter restene av humant BAFF. I et annet aspekt er BAFFantagonisten et BR3-antistoff. Et «BR3-antistoff» er et antistoff som binder BR3, og fortrinnsvis binder BR3 innenfor en region av humant BR3 som omfatter restene
68 av humant BR3. Sekvensene til humant BAFF og humant BR3 er funnet f.eks. i US 2006/ Andre eksempler på BAFF-bindende polypeptider eller BAFF-antistoffer kan finnes f.eks. i WO 2002/092620, WO 2003/014294, Gordon et al., Biochemistry 42(20): (2003), Kelley et al., J. Biol.Chem. 279: (2004), WO 1998/18921, WO 2001/12812, WO 2000/68378 og WO 2000/ Et «antistoff mot IgE» inkluderer alle antistoffer som binder IgE spesifikt på en slik måte at det ikke utløser tverrbinding når IgE bindes til høyaffinitetsreseptoren på mastceller og basofiler. Eksempler på antistoffer inkluderer antistoffene i oppfinnelsen, samt rhumabe2- (E2, XOLAIR ), E26-, E27- og CGP-1- (Hu- 901), samt HA-antistoffet. Aminosyresekvensene til de tunge og lette kjedevariable domenene til de humaniserte antistoffene mot IgE E2, E26 og E27 er offentliggjort for eksempel i U.S.P og WO99/016. CGP-1-(Hu-901)-antistoffet er beskrevet i Corne et al., (1997) J. Clin. Invest. 99(): , WO 92/17207 og ATCC-dep. nr. BRL-706, BRL-11130, BRL-11131, BRL og BRL HAantistoffet er beskrevet i USSN 60/444,229, WO2004/ og WO2004/0700. II. Måter å utføre oppfinnelsen på 20 A. Rekombinant fremstilling Oppfinnelsen tilveiebringer også en isolert nukleinsyre som koder for apoptotiske antistoffer mot IgE, vektorer og vertsceller som omfatter denne nukleinsyren og rekombinante metoder for å fremstille antistoffet. For rekombinant fremstilling av antistoffet blir nukleinsyren som koder for det isolert og satt inn i en replikerbar vektor for ytterligere kloning (forsterkning av DNA-et) eller for uttrykking. DNA som koder for monoklonalt antistoff isoleres og sekvenseres enkelt ved å anvende konvensjonelle prosedyrer (for eksempel ved hjelp av oligonukleotidsonder som kan bindes spesifikt til gener som koder for de tunge og lette kjedene til antistoffet). Mange vektorer er tilgjengelige. Vektorkomponentene inkluderer generelt, men er ikke begrenset til, én eller flere av de følgende: en signalsekvens, et replikasjonsorigo, ett eller flere markørgener, og forsterkerelement, en promotor og en transkripsjonsstoppsekvens. (1) Signalsekvenskomponenten
69 67 1 Antistoffene mot IgE ifølge denne oppfinnelsen kan fremstilles rekombinant, ikke bare direkte, men også som et fusjonspolypeptid med et heterologt polypeptid som fortrinnsvis er en signalsekvens eller et annet polypeptid som har et spesifikt spaltingssete i N-enden av det modne proteinet eller polypeptidet. Den valgte heterologe signalsekvensen er fortrinnsvis en som gjenkjennes og behandles (dvs. spaltes med en signalpeptidase) av vertscellen. For prokaryotisk vertsceller som ikke gjenkjenner og behandler den naturlige pattedyrsignalsekvensen, erstattes signalsekvensen med en prokaryotisk signalsekvens valgt f.eks. fra gruppen alkaliske fosfatase, penicillinase, lpp eller varmestabile enterotoksin-iilederpeptider. For gjærutskilling kan den opprinnelige signalsekvensen erstattes for eksempel med gjærinvertaseleder, faktorleder (inkludert Saccharomyces- og Kluyveromycesfaktorledere), eller syrefosfataseleder, C. albicansglukoamylaseleder eller signalpeptidet som er beskrevet i WO 90/ Når det gjelder pattedyrcelleuttrykkingen er det tilgjengelig både pattedyrsignalsekvenser og virale sekretoriske ledere, for eksempel herpes simplex gd-signalpeptidet. DNA-et for en slik forløperregion bindes i leserammen til DNA som koder for det IgE-bindende antistoffet. 20 (2) Replikasjonsorigo 2 30 Både uttrykkings- og kloningsvektorer inneholder en nukleinsyresekvens som gjør det mulig for vektoren å replikere i én eller flere utvalgte vertsceller. I kloningsvektorer er denne sekvensen vanligvis en sekvens som gjør det mulig for vektoren å replikere uavhengig av vertens kromosom-dna, og inkluderer replikasjonsorigoer eller autonomt replikerende sekvenser. Slike sekvenser er godt kjent for en rekke bakterier, gjærceller og virus. Replikasjonsorigoen fra plasmidet pbr322 er egnet for de fleste Gram-negative bakteriene, plasmidorigoen på 2 µ er egnet for gjær og forskjellige virusorigoer (SV40, polyom, adenovirus, VSV eller BPV) er nyttige for kloningsvektorer i pattedyrceller. Generelt er replikasjonsorigokomponenten ikke nødvendig for pattedyruttrykkingsvektorer (SV40-origoen kan vanligvis anvendes bare fordi den inneholder den tidlige promotoren). 3 (3) Seleksjonsgenkomponenten
70 68 Uttrykkings- og kloningsvektorer kan inneholde et seleksjonsgen, også kalt en selekterbar markør. Typiske seleksjonsgener koder for proteiner som (a) gir resistens mot antibiotika eller andre toksiner, f.eks. ampicillin, neomycin, metotreksat eller tetrasyklin, (b) komplementerer auksotrofe mangler, eller (c) tilfører viktige næringsstoffer som ikke er tilgjengelige fra komplekst medium, f.eks. genet som koder for D-alaninrasemase for Bacilli. I ett eksempel på et seleksjonsskjema anvendes det et legemiddel til å stanse veksten av en vertscelle. De cellene som transformeres vellykket med et heterologt gen, produserer et protein som gir legemiddelresistens og dermed overlever seleksjonsregimet. I eksempler på slik dominant seleksjon anvendes legemidlene neomycin, mykofenolsyre og hygromycin. 1 Et annet eksempel på egnede selekterbare markører for pattedyrceller er slike som gjør det mulig å identifisere celler som er kompetente til å ta opp den IgEbindende antistoffnukleinsyren, som f.eks. DHFR, tymidinkinase, metallotionein-i og -II, fortrinnsvis primatgener for metallotionein, adenosindeaminase, ornitindekarboksylase osv For eksempel identifiseres celler med DHFR-seleksjonsgen først ved å dyrke alle transformantene i et dyrkingsmedium som inneholder metotreksat (Mtx), en kompetitiv antagonist for DHFR. Når villtype-dhfr anvendes, er en egnet vertscelle den eggstokkcellestammen fra kinesisk dverghamster (CHO) som mangler DHFR-aktivitet (f.eks. ATCC CRL-9096). Alternativt kan vertsceller (særlig villtypeverter som inneholder endogen DHFR) transformeres eller kotransformeres med DNA-sekvenser som koder for IgEbindende antistoff, villtype-dhfr-protein og en annen selekterbar markør, som for eksempel aminoglykosid-3'-fosfotransferase (APH) av cellevekst i medium som inneholder et seleksjonsmiddel for den selekterbare markøren, som f.eks. et aminoglykosidisk antibiotikum, f.eks. kanamycin, neomycin eller G418. Se U.S. patent nr Et egnet seleksjonsgen for anvendelse i gjær er trp1-genet som finnes i gjærcelleplasmidet YRp7 (Stinchcomb et al., Nature, 282:39 (1979)). Trp1-genet gir en seleksjonsmarkør for en mutert stamme av gjærceller som mangler evnen til å vokse i tryptofan, for eksempel ATCC nr eller PEP4-1. Jones,
71 69 Genetics, 8:12 (1977). Nærværet av trp1-lesjonen i vertscellegenomet gir da et effektivt miljø for å registrere transformering ved vekst i fravær av tryptofan. Tilsvarende suppleres de Leu2-manglende gjærstammene (ATCC eller ) ved hjelp av kjente plasmider som bærer Leu2-genet. I tillegg kan vektorer som avledes fra det sirkulære plasmidet pkd1 på 1,6 µm anvendes til å transformere Kluyveromyces-gjærceller. Alternativt er et uttrykkingssystem for storskalaproduksjon av rekombinant kalvekymosin rapportert for K. lactis. Van den Berg, Bio/Technology, 8:13 (1990). Det er også offentliggjort stabile vektorer for flerkopiuttrykking for utskilling av modent rekombinant humant serumalbumin fra industrielle stammer av Kluyveromyces Fleer et al., Bio/Technology, 9: (1991) (4) Promotorkomponenten Uttrykkings- og kloningsvektorer inneholder vanligvis en promotor som gjenkjennes av vertsorganismen, og er operativt koblet til nukleinsyren som koder for det IgE-bindende antistoffet. Promotorer som er egnet for anvendelse med prokaryotiske verter inkluderer phoa- promoteren, laktamase- og laktosepromotorsystemer, alkalisk fosfatasepromotor, et tryptofan-(trp)- promotorsystem og hybride promoterer som f.eks. tac-promotoren. Men andre kjente bakteriepromotorer er også egnet. Promotorer for anvendelse i bakteriesystemer vil også inneholde en Shine-Dalgarno-(S.D.)-sekvens operativt koblet til DNA-et som koder for det IgE-bindende antistoffet. Promotorsekvenser er kjent for eukaryoter. Nesten alle eukaryotiske gener har en AT-rik region som ligger omtrent 2 til 30 baser oppstrøms fra setet transkriberingen starter fra. En annen sekvens som finnes 70 til 80 baser oppstrøms fra transkriberingsstarten til mange gener er en CNCAAT-region der N kan være et hvilket som helst nukleotid. Ved 3'-enden av de fleste eukaryotiske gener er det en AATAAA-sekvens som kan være signalet for å tilsette poly-a-halen til 3'-enden av den kodingssekvensen. Alle disse sekvensene kan hensiktsmessig settes inn i eukaryotiske uttrykkingsvektorer. 3 Eksempler på egnede promotorsekvenser for anvendelse med gjærverter inkluderer promotorene for 3-fosfoglyseratkinase eller andre glykolytiske enzymer som f.eks. enolase, glyseraldehyd-3-fosfatedehydrogenase,
72 70 heksokinase, pyruvatdekarboksylase, fosfofruktokinase, glukose-6-fosfatisomerase, 3-fosfoglyseratmutase, pyruvatkinase, triosefosfatisomerase, fosfoglukoseisomerase og glukokinase. Andre gjærpromotorer, som er induserbare promotorer med den ytterligere fordelen at transkriberingen kontrolleres av vekstforholdene, er promotorregionene for alkoholdehydrogenase 2, isocytokrom C, sur fosfatase, nedbrytende enzymer forbundet med nitrogenmetabolisme, metallotionein, glyseraldehyd-3-fosfatdehydrogenase og enzymer som er ansvarlige for maltoseog galaktoseutnyttelsen. Egnede vektorer og promotorer for anvendelse i gjærcelleuttrykkingen er også beskrevet i EP 73,67. Gjærcelleforsterkere anvendes også med fordel sammen med gjærcellepromotorer IgE-bindende antistofftranskribering fra vektorer i pattedyrvertsceller kontrolleres, for eksempel av promotorer fra genomene til virus som f.eks. polyomvirus, fuglekoppervirus, adenovirus (som f.eks. adenovirus 2), bovint papillomavirus, aviært sarkomvirus, cytomegalovirus, et retrovirus, hepatitt B-virus og mest foretrukket Simian Virus 40 (SV40), fra heterologe pattedyrpromotorer, f.eks. aktinpromotoren eller en immunoglobulinpromotor, fra varmesjokkpromotorer, forutsatt at slike promotorer er kompatible med vertscellesystemene De tidlige og sene promotorene for SV40-viruset fås beleilig som et SV40- restriksjonsfragment som også inneholder SV40-virusreplikasjonsorigoen. Den umiddelbare tidlige promotoren fra det humane cytomegaloviruset fås beleilig som et Hindlll E-restriksjonsfragment. Et system for å uttrykke DNA i pattedyrverter med det bovine papillomaviruset som en vektor er offentliggjort i U.S. patent nr En modifikasjon av dette systemet er beskrevet i U.S. patent nr Se også Reyes et al., Nature 297: (1982) om uttrykking av humant interferon cdna i museceller under kontroll av en tymidinkinasepromotor fra herpes simplex-virus. Alternativt kan den lange repeterende enden fra Rous sarkomavirus anvendes som promotor. () Forsterkerelementkomponenten 3 Høyere eukaryoters transkribering av et DNA som koder for det IgE-bindende antistoffet ifølge denne oppfinnelsen økes ofte ved å sette inn en forsterkersekvens i vektoren. Det er nå kjent mange forsterkersekvenser fra
73 71 pattedyrgener (globin, elastase, albumin, α-fetoprotein og insulin). Man vil imidlertid vanligvis anvende en forsterker fra et virus for eukaryotiske celler. Eksempler inkluderer SV40-forsterkeren på den sene siden av replikasjonsorigoen (bp 0 270), den tidlige promotorforsterkeren for cytomegalovirus, polyomforsterkeren på den sene siden av replikasjonsorigoen og adenovirusforsterkere. Se også Yaniv, Nature 297:17-18 (1982) om forsterkende elementer for aktivering av eukaryotiske promotorer. Forsterkeren kan spleises inn i vektoren i en posisjon ' eller 3' til den IgEbindende antistoffkodingssekvensen, men plasseres fortrinnsvis i en -posisjon fra promotoren. (6) Transkripsjonsstoppkomponenten 1 20 Uttrykkingsvektorer som anvendes i eukaryotiske vertsceller (gjær, sopp, insekt, plante, dyr, menneske eller kjerneceller fra andre flercellede organismer) vil også inneholde sekvenser som er nødvendige for å avslutte transkriberingen og for å stabilisere mrna-et. Slike sekvenser kan vanligvis fås fra utranslaterte '- og av og til 3 -regioner av eukaryotisk eller viralt DNA eller cdna. Disse regionene inneholder nukleotidsegmenter transkribert som polyadenylerte fragmenter i den utranslaterte delen av mrna-et som koder for IgE-bindende antistoff. En nyttig komponent for transkripsjonsstopp er polyadenyleringsregionen av det bovine veksthormonet. Se WO94/126 og utrykkingsvektoren beskrevet der (7) Valg og transformering av vertsceller Egnede vertsceller for kloning eller uttrykking av DNA-et i vektorene i dette dokumentet er de prokaryotiske cellene, gjærcellene og høyere eukaryotiske cellene som beskrives ovenfor. Egnede prokaryoter for dette formålet inkluderer eubakterier, som f.eks. Gram-negative eller Gram-positive organismer, f.eks. Enterobacteriaceae, som f.eks. Escherichia, f.eks. E. coli, Enterobacter, Erwinia, Klebsiella, Proteus, Salmonella, f.eks. Salmonella typhimurium, Serratia, f.eks. Serratia marcescans og Shigella, så vel som Bacilli som f.eks. B. subtilis og B. licheniformis (f.eks. B. licheniformis 41P offentliggjort i DD 266,7 publisert 12. april 1989), Pseudomonas, som feks. P. aeruginosa og Streptomyces. En foretrukket E. coli-kloningsvert er E. coli 294 (ATCC 31,446), men andre stammer som f.eks. E. coli B, E. coli X1776 (ATCC 31,37) og E. coli W31 (ATCC 27,32) er egnet. Disse eksemplene er illustrerende og ikke begrensende.
74 Fullengdeantistoff, antistoffragmenter og antistoffusjonsproteiner kan produseres i bakterier, særlig når glykosylering og Fc-effektorfunksjon ikke er nødvendig, som f.eks. når det terapeutiske antistoffet konjugeres til et cytotoksisk middel (f.eks. et toksin) og immunkonjugatet i seg selv viser effektivitet ved ødeleggelse av tumorceller. Fullengdeantistoffer har større halveringstid i sirkulasjonen. Produksjonen i E. coli er raskere og mer kostnadseffektiv. Om uttrykking av antistoffragmenter og polypeptider i bakterier, se f.eks. U.S (Carter et. al.), U.S (Joly et al.) og U.S (Simmons et al.) som beskriver translasjonsstartregionen (TIR) og signalsekvenser for å optimalisere uttrykking og utskilling. Etter uttrykkingen isoleres antistoffet fra E. coli-cellepastaen i en løselig fraksjon og kan renses ved f.eks. en protein A- eller G-kolonne avhengig av isotypen. Endelig rensing kan utføres tilsvarende prosessen for rensing av antistoffet som uttrykkes for eksempel i CHO-celler. I tillegg til prokaryoter er eukaryotiske mikrober som f.eks. trådformet sopp eller gjærceller egnede klonings- eller uttrykkingsverter for IgE-bindende antistoffkodende vektorer. Saccharomyces cerevisiae, eller vanlig bakegjær, er den som anvendes hyppigst av de lavere eukaryotiske vertsmikroorganismene. En rekke andre slekter, arter og stammer er imidlertid lett tilgjengelige og kan anvendes ifølge dette dokumentet, som f.eks. Schizosaccharomyces pombe; Kluyveromyces-verter som f.eks. K. lactis, K. fragilis (ATCC ), K. bulgaricus (ATCC 16 04), K. wickeramii (ATCC ), K. waltii (ATCC 6 00), K. drosophilarum (ATCC ), K. thermotolerans og K. marxianus; yarrowia (EP ); Pichia pastoris (EP ); Candida; Trichoderma reesia (EP ); Neurospora crassa; Schwanniomyces, som f.eks. Schwanniomyces occidentalis; og trådformet sopp som f.eks. Neurospora, Penicillium, Tolypocladium og Aspergillus-verter som f.eks. A. nidulans og A. niger Egnede vertsceller for uttrykking av glykosylert IgE-bindende antistoff avledes fra flercellede organismer. Eksempler på virvelløse celler inkluderer plante- og insektsceller. Tallrike bakulovirusstammer og varianter og tilsvarende betingede insektvertsceller fra verter som f.eks. Spodoptera frugiperda (larve), Aedes aegypti (mygg), Aedes albopictus (mygg), Drosophila melanogaster (bananflue) og Bombyx mori er identifisert. En rekke virusstammer for transfektering er tilgjengelige for offentligheten, for eksempel kan L-1-varianten til Autographa californica NPV- og Bm--stammen til Bombyx mori NPV, og slike virus anvendes
75 73 som viruset i dette dokumentet ifølge den foreliggende oppfinnelsen, særlig for transfektering av Spodoptera frugiperda-celler. Plantecellekulturer av bomull, mais, potet, soyabønne, petunia, tomat og tobakk kan også benyttes som verter Interessen har imidlertid vært størst for virveldyrceller, og formering av virveldyrceller i kultur (vevskultur) har blitt en rutineprosedyre. Eksempler på nyttige pattedyrvertscellestammer er CV1-stammen fra apenyrer transformert med SV40 (COS-7, ATCC CRL 161); den humane fosternyrestammen (293 eller 293-celler subklonet for vekst i suspensjonskultur, Graham et al., J. Gen Virol. 36:9 (1977)); babyhamsternyreceller (BHK, ATCC CCL ); eggstokkceller fra kinesisk dverghamster/-dhfr (CHO, Urlaub et al., Proc. Natl. Acad. Sci. USA 77:4216 (1980)); murine sertoliceller (TM4, Mather, Biol. Reprod. 23: (1980)); apenyreceller (CV1 ATCC CCL 70); nyreceller fra grønn marekatt (VERO-76, ATCC CRL-187); humane livmorhalskarsinomceller (HELA, ATCC CCL 2); hundenyreceller (MDCK, ATCC CCL 34); bøffelrotteleverceller (BRL 3A, ATCC CRL 1442); humane lungeceller (W138, ATCC CCL 7); humane leverceller (Hep G2, HB 806); murine spenetumorceller (MMT 06062, ATCC CCL1); TRI-celler Mather et al., Annals N.Y. Acad. Sci. 383:44 68 (1982)); MRC -celler; FS4- celler; og en human hepatomlinje (Hep G2). 2 Vertsceller transformeres med de ovenfor beskrevne uttrykkings- eller kloningsvektorene for IgE-bindende antistoffproduksjon og dyrkes i konvensjonelle næringsmedier med egnede modifikasjoner for å indusere promotorer, velge transformanter eller forsterke genene som koder for de ønskede sekvensene. (8) Dyrking av vertscellene 30 3 Vertscellene som anvendes til å produsere IgE-bindende antistoff ifølge denne oppfinnelsen kan dyrkes i en rekke medier. Kommersielt tilgjengelige medier som Hams F (Sigma), minimalt essensielt medium ((MEM), (Sigma), RPMI-1640 (Sigma) og Dulbeccos modifiserte Eagles medium ((DMEM), Sigma) er egnet for dyrking av vertscellene. I tillegg kan et hvilket som helst av mediene som beskrives i Ham et al., Meth. Enz. 8:44 (1979), Barnes et al., Anal. Biochem.2:2 (1980), U.S. pat. nr ; ; ; ; eller ; WO 90/03430; WO 87/0019; eller U.S. Patent Re anvendes som
76 74 dyrkingsmedium for vertscellene. Et hvilket som helst av disse mediene kan suppleres etter behov med hormoner og/eller andre vekstfaktorer (som f.eks. insulin, transferrin eller epidermal vekstfaktor), salter (som f.eks. natriumklorid, kalsium, magnesium og fosfat), buffere (som f.eks. HEPES), nukleotider (som f.eks. adenosin og tymidin), antibiotika (som f.eks. GENTAMYCIN -legemiddel), sporstoffer (definert som uorganiske forbindelser som vanligvis er til stede i sluttkonsentrasjoner i det mikromolare området), og glukose eller en tilsvarende energikilde. Eventuelle andre nødvendige tilskudd kan også inkluderes ved passende konsentrasjoner som vil være kjent for fagfolk. Dyrkningsforholdene, som f.eks. temperatur, ph og lignende, er slike som tidligere er anvendt med vertscellen for uttrykking, og vil være åpenbare for fagfolk med vanlige ferdigheter. (9) Rensing av antistoffet Ved å anvende rekombinante metoder kan antistoffet fremstilles intracellulært, i det periplasmiske rommet eller direkte skilt ut i mediet. Dersom antistoffet fremstilles intracellulært, som et første trinn, fjernes de partikkelformede restene, enten vertsceller eller lyserte fragmenter, for eksempel ved sentrifugering eller ultrafiltrering. Carter et al., Bio/Technology : (1992) beskriver en prosedyre for å isolere antistoffer som utskilles i det periplasmiske rommet til E. coli. Kort fortalt tines cellepasta i nærvær av natriumacetat (ph 3,), EDTA og fenylmetylsulfonylfluorid (PMSF) i løpet av rundt 30 min. Cellerestene kan fjernes ved sentrifugering. Der antistoffet skilles ut i mediet, blir supernatantene fra slike uttrykkingssystemer generelt først konsentrert ved å anvende et kommersielt tilgjengelig proteinkonsentrasjonsfilter, for eksempel en Amicon- eller Millipore Pellicon-ultrafiltreringsenhet. En proteasehemmer som f.eks. PMSF kan inkluderes i en hvilken som helst av de foregående trinnene for å hemme proteolysen, og antibiotika kan inkluderes for å forhindre vekst av tilfeldige forurensninger Antistoffsammensetningen som fremstilles fra cellene kan renses ved hjelp av f.eks. hydroksylapatittkromatografi, gelelektroforese, dialyse og affinitetskromatografi, med affinitetskromatografi som den foretrukne rensemetoden. Egnetheten til protein A som en affinitetsligand avhenger av artene og isotypen til eventuelle immunoglobulin Fc-domener som er til stede i antistoffet. Protein A kan anvendes til å rense antistoff basert på 1, 2 eller 4 humane tunge kjeder (Lindmark et al., J. Immunol. Meth. 62:1 13 (1983)). Protein G er anbefalt for alle museisotyper og for humant 3 (Guss et al., EMBO J. :16717 (1986)).
77 7 Matriksen affinitetsliganden festes til er som oftest agarose, men andre matrikser er tilgjengelige. Mekanisk stabile matrikser, som f.eks. glass med kontrollert porediameter eller poly(styren-divinyl)benzen tillater raskere strømningshastighet og kortere behandlingstid enn det som kan oppnås med agarose. Hvis antistoffet omfatter et C H 3-domene er Bakerbond ABX -harpiksen (J. T. Baker, Phillipsburg, NJ) nyttig for rensing. Andre metoder for proteinrensing som f.eks. fraksjonering på en ionebytterkolonne, etanolfelling, HPLC med omvendte faser, kromatografi på silika, kromatografi på heparin-sepharose -kromatografi på en anion- eller kationbyttermasse (f.eks. en polyasparaginsyrekolonne), kromatofokusering, SDS-PAGE og ammoniumsulfatfelling er også tilgjengelig avhengig av antistoffet som skal gjenvinnes. 1 Etter et eventuelt innledende rensetrinn, kan blandingen som omfatter antistoffet av interesse og forurensninger utsettes for kromatografi med hydrofobe interaksjoner og lav ph ved hjelp av en elueringsbuffer med ph mellom omtrent 2, og 4,, fortrinnsvis utført ved lav saltkonsentrasjon (f.eks. fra rundt 0 0,2 M salt). B. Antistoffremstilling 20 1) Polyklonale antistoffer Polyklonale antistoffer rekrutteres generelt i dyr med flere subkutane (sc) eller intraperitoneale (ip) injeksjoner av det relevante antigenet og en adjuvans. Det kan være nyttig å konjugere det relevante antigenet til et protein som er immunogent i artene som skal immuniseres, f.eks. keyhole limpet hemocyanin (KLH), serumalbumin, bovint tyroglobulin eller soyabønnetrypsinhemmer, ved å anvende et bifunksjonelt eller derivatiserende middel, f.eks. maleimidobenzoylsulfosuksinimidester (konjugering gjennom cysteinrester), N- hydroksysuksinimid (gjennom lysinrester), glutaraldehyd, ravsyreanhydrid, SOCl 2 eller R 1 N=C=NR, der R og R 1 hver for seg er lavere alkylgrupper. Eksempler på adjuvanser som kan benyttes inkluderer Freunds komplette adjuvans og MPL-TDM-adjuvans (monofosforyllipid A, syntetisk trehalosedikorynomykolat). Immuniseringsprotokollen kan velges av en fagmann på området uten unødig eksperimentering. Dyrene immuniseres mot antigenet, immunogene konjugater eller derivater ved å kombinere for eksempel 0 µg eller µg av proteinet eller konjugatet (for
78 76 henholdsvis kaniner eller mus) med 3 volum av Freunds komplette adjuvans og injisere løsningen intradermalt flere steder. En måned senere får dyrene boosterinjeksjoner med 1/ til 1/ av den opprinnelige mengden peptid eller konjugat i Freunds komplette adjuvans ved subkutan injeksjon flere steder. Sju til fjorten dager senere blir dyrene tappet for blod og serumet analysert på antistofftiter. Dyrene får boosterinjeksjoner inntil titeren flater ut. Konjugatene kan også lages i rekombinant cellekultur som proteinfusjoner. Videre er aggregerende midler som f.eks. alun egnet til å forbedre immunresponsen. 2) Monoklonale antistoffer 1 Monoklonale antistoffer fås fra en populasjon av stort sett homogene antistoffer, dvs. at de individuelle antistoffene som omfatter populasjonen er identiske med unntak av mulige naturlig forekommende mutasjoner og/eller posttranslasjonelle modifikasjoner (f.eks. isomerisering, amidering) som kan være til stede i mindre mengder. Altså sier adjektivet «monoklonal» at antistoffet ikke er en blanding av adskilte antistoffer. 20 For eksempel kan de monoklonale antistoffene lages ved å anvende hybridommetoden som først beskrives av Kohler et al., Nature, 26:49 (197), eller kan lages ved rekombinant DNA-metoder (U.S. patent nr ) I hybridommetoden blir en mus eller et annet passende vertsdyr, som f.eks. en hamster, immunisert som beskrevet ovenfor i dette dokumentet for å fremkalle lymfocytter som produserer eller kan produsere antistoffer som vil binde spesifikt til proteinet som anvendes til immuniseringen. Lymfocyttene kan alternativt immuniseres in vitro. Lymfocyttene fusjoneres deretter med myelomceller ved å anvende et egnet fusjoneringsmiddel, som f.eks. polyetylenglykol, til å danne en hybridomcelle (Goding, Monoclonal Antibodies: Principles and Practice, s. 9 3 (Academic Press, 1986). 3 Immuniseringsmidlet vil typisk inkludere det antigene proteinet eller en fusjonsvariant av dette. Generelt anvendes enten lymfocytter fra perifert blod («PBL-er») dersom celler av human opprinnelse er ønsket, eller miltceller eller lymfeknuteceller dersom ikke-humane pattedyrkilder er ønsket. Lymfocyttene fusjoneres deretter med en udødeliggjort cellestamme ved hjelp av et egnet
79 77 fusjoneringsmiddel, f.eks. polyetylenglykol, for å danne en hybridomcelle. Goding, Monoclonal Antibodies: Principles and Practice, Academic Press (1986), s Udødeliggjorte cellestammer er vanligvis transformerte pattedyrceller, særlig myelomceller fra gnagere, storfe og mennesker. Det benyttes vanligvis rotteeller musemyelomcellestammer. Hybridomcellene som fremstilles på denne måten blir utsådd og dyrket i et egnet kulturmedium som fortrinnsvis inneholder ett eller flere stoffer som hemmer veksten eller overlevelsen av de ufusjonerte, opphavsmyelomcellene. Hvis f.eks. opphavsmyelomcellene mangler enzymet hypoxantinguaninfosforibosyltransferase (HGPRT eller HPRT), vil kulturmediet for hybridomene typisk inkludere hypoxantin, aminopterin og tymidin (HATmedium), som er stoffer som forhindrer veksten av HGPRT-manglende celler Foretrukne udødeliggjorte myelomceller er slike som fusjonerer effektivt, støtter stabilt høy produksjon av antistoff i de valgte antistoffproduserende cellene, og er følsomme for et medium, som f.eks. et HAT-medium. Blant disse foretrekkes de murine myelomlinjene, f.eks. de som avledes fra MOPC-21- og MPC-11-musetumorer som er tilgjengelige fra Salk Institute Cell Distribution Center, San Diego, California USA og SP-2-celler (og derivater av disse, f.eks. X63-Ag8-63) tilgjengelig fra the American Type Culture Collection, Manassas, Virginia USA. Humane myelom- og murine-humane heteromyelomcellestammer er også beskrevet for fremstilling av humane monoklonale antistoffer (Kozbor, J. Immunol., 133:3001 (1984); Brodeur et al., Monoclonal Antibody Production Techniques and Applications, s (Marcel Decker, Inc., New York, 1987)). Kulturmedium der det vokser hybridomceller analyseres for produksjon av monoklonale antistoffer mot antigenet. Fortrinnsvis bestemmes bindingsspesifisiteten til monoklonale antistoffer som produseres av hybridomceller ved immunutfelling eller ved en in vitro bindingsanalyse, f.eks. radioimmunanalyse (RIA) eller enzymbundet immunsorbentanalyse (ELISA). 3 Kulturmediet der hybridomcellene dyrkes kan analyseres for nærvær av monoklonale antistoffer mot det ønskede antigenet. Fortrinnsvis kan bindingsaffiniteten og bindingsspesifisiteten til det monoklonale antistoffet bestemmes ved immunutfelling eller ved en in vitro bindingsanalyse, som f.eks. radioimmunanalyse (RIA) eller enzymbundet analyse (ELISA). Slike metoder og
80 78 analyser er kjent i faget. For eksempel kan bindingsaffiniteten bestemmes ved Scatchard-analysen til Munson et al., Anal. Biochem., 7:220 (1980) Etter at det er identifisert hybridomceller som produserer antistoffer med den ønskede spesifisiteten, affiniteten og/eller aktiviteten, kan klonene subklones ved begrensende fortynningsprosedyrer og dyrkes ved standardmetoder (Goding, ovenfor). Egnede dyrkingsmedier for dette formålet inkluderer for eksempel D-MEM- eller RPMI-1640-medium. I tillegg kan hybridomcellene dyrkes in vivo som tumorer i et pattedyr. De monoklonale antistoffene som skilles ut av subklonene separeres fra dyrkingsmediet, ascitesvæsken eller serum ved konvensjonelle renseprosedyrer for immunoglobulin som for eksempel protein A-sefarose, hydroksylapatittkromatografi, gelelektroforese, dialyse eller affinitetskromatografi. Monoklonale antistoffer kan også fremstilles ved rekombinant DNA-metoder, f.eks. slike som er beskrevet i U.S. patent nr , og som beskrevet ovenfor. DNA som koder for de monoklonale antistoffene isoleres og sekvenseres enkelt ved konvensjonelle prosedyrer (f.eks. ved å anvende oligonukleotidsonder som kan bindes spesifikt til gener som koder for de tunge og lette kjedene av murine antistoffer). Hybridomcellene tjener som en foretrukket kilde for slikt DNA. Når det er isolert, kan DNA plasseres i uttrykkingsvektorer, som deretter transfekteres inn i vertsceller, som for eksempel E. coli-celler, ape-cos-celler, eggstokkceller fra kinesisk dverghamster (CHO) eller myelomceller som ikke på annen måte produserer immunoglobulinprotein, for å syntetisere monoklonale antistoffer i slike rekombinante vertsceller. Oversiktsartikler om rekombinant uttrykking av DNA som koder for antistoffet i bakterier inkluderer Skerra et al., Curr. Opinion in Immunol., : (1993) og Plückthun, Immunol. Revs. 130: (1992) I en annen utførelsesform kan antistoffer isoleres fra bakteriofagbibliotek av antistoffer som lages ved hjelp av metodene som er beskrevet i McCafferty et al., Nature, 348:2 4 (1990). Clackson et al., Nature, 32: (1991) og Marks et al., J. Mol. Biol., 222:81 97 (1991) beskriver isolering av henholdsvis murine og humane antistoffer ved hjelp av bakteriofagbibliotek. Senere publikasjoner beskriver fremstilling av humane antistoffer med høy affinitet (nm-området) ved kjedeomstokking (Marks et al., Bio/Technology, : (1992)), og kombinatorisk infektering og in vivo-rekombinering
81 79 1 som en strategi for å konstruere meget store bakteriofagbibliotek (Waterhouse et al., Nucl. Acids Res., 21: (1993)). Altså er disse metodene levedyktige alternativer til tradisjonelle monoklonale antistoffhybridommetoder for isolering av monoklonale antistoffer. DNA-et kan også modifiseres, for eksempel ved å erstatte de homologe murine sekvensene med sekvensen som koder for humane konstante domener med tung og lett kjede (U.S. patent nr ; Morrison, et al., Proc. Natl Acad. Sci. USA, 81:681 (1984)), eller ved kovalent sammenføying av hele eller deler av kodingssekvensen for et ikke-immunoglobulinpolypeptid til kodingssekvensen for immunoglobulin. Typisk byttes de konstante domenene til et antistoff ut med slike ikke-immunoglobulinpolypeptider, eller de variable domenene til et antigenkombinerende sete av et antistoff byttes ut med dem for å lage et kimært toverdig antistoff som omfatter et antigenkombinerende sete som har spesifisitet for et antigen og et annet antigenkombinerende sete som har spesifisitet for et annet antigen De monoklonale antistoffene som er beskrevet i dette dokumentet kan være enverdige, og fremstillingen av disse er godt kjent i faget. For eksempel involverer en metode rekombinant uttrykking av den lette kjeden av immunoglobulinet og en modifisert tung kjede. Den tunge kjeden trunkeres generelt på et hvilket som helst punkt i Fc-regionen, for å hindre kryssbinding av den tunge kjeden. Alternativt kan de aktuelle cysteinrestene byttes ut med en annen aminosyrerest eller fjernes, for å hindre kryssbinding. In vitro-metoder er også egnet for fremstilling av enverdige antistoffer. Antistoffer kan digereres for å lage fragmenter av dem, særlig Fabfragmenter, ved rutinemessige metoder som er kjent i faget. 30 Kimære eller hybride antistoffer kan også fremstilles in vitro ved kjente metoder innen syntetisk proteinkjemi, inkludert slike som involverer kryssbindingsmidler. For eksempel kan immuntoksiner konstrueres ved hjelp av en disulfidutvekslingsreaksjon eller ved å danne en tioeterbinding. Eksempler på egnede reagenser for dette formålet inkluderer iminotiolat og metyl-4-merkaptobutyrimidat. 3 3) Humaniserte antistoffer. Antistoffene ifølge oppfinnelsen kan også omfatte humaniserte eller humane antistoffer. Humaniserte former av ikke-humane (for eksempel murine)
82 antistoffer er kimære immunoglobuliner, immunoglobulinkjeder eller fragmenter av disse (slik som Fv, Fab, Fab', F(ab') 2 eller andre antigenbindende undersekvenser av antistoffer) som inneholder minimalt med sekvens fra ikke-humant immunoglobulin. Humaniserte antistoffer inkluderer humane immunoglobuliner (mottakerantistoff) der aminosyrerester fra en komplementaritetsbestemmende region (CDR) i mottakeren byttes ut med rester fra en CDR fra en ikke-human art (donorantistoff) som f.eks. mus, rotte eller kanin som har den ønskede spesifisiteten, affiniteten og kapasiteten. I noen tilfeller byttes Fv-rammerester av det humane immunoglobulinet ut med tilsvarende ikke-humane rester. Humaniserte antistoffer kan også omfatte rester som ikke finnes verken i mottakerantistoffet eller i de importerte CDRene eller rammesekvensene. Generelt vil det humaniserte antistoffet omfatte i det vesentlige alt av minst ett, og typisk to variable domener, der alle eller i det vesentlige alle CDR-regionene tilsvarer regionene til et ikke-humant immunoglobulin, og alle eller i det vesentlige alle FR-regionene er regioner fra en human immunoglobulinsekvens. Optimalt vil det humaniserte antistoffet også omfatte minst en del av en konstant immunoglobulinregion (Fc), vanligvis den konstante regionen til et humant immunoglobulin. Jones et al., Nature 321: 22 2 (1986); Riechmann et al., Nature 332: (1988) og Presta, Curr. Opin. Struct. Biol. 2: (1992) Fremgangsmåter for å humanisere ikke-humane antistoffer er godt kjente i faget. Generelt har et humanisert antistoff én eller flere aminosyrerester som er innført i det fra en ikke-human kilde. Disse ikke-humane aminosyrerestene blir ofte omtalt som «import»-rester som vanligvis tas fra et variabelt «import»-domene. Humaniseringen kan utføres i det vesentlige ved å følge metoden til Winter og hans medarbeidere, Jones et al., Nature 321:22 2 (1986); Riechmann et al., Nature 332: (1988); Verhoeyen et al., Science 239: (1988), eller ved å erstatte de tilsvarende sekvensene i et humant antistoff med gnager-cdr eller CDR-sekvenser. Følgelig er slike «humaniserte» antistoffer kimære antistoffer (U.S. patent nr ), der betydelig mindre enn et intakt humant variabelt domene er erstattet med den tilsvarende sekvensen fra en ikke-human art. I praksis er humaniserte antistoffer vanligvis humane antistoffer der noen CDR-rester og muligens noen FR-rester byttes ut med rester fra analoge posisjoner i gnagerantistoffer.
83 81 Valget av humane variable domener, både lette og tunge, som skal anvendes i fremstillingen av de humaniserte antistoffene er svært viktig for å redusere antigenisiteten. Ifølge den såkalte «beste tilpasnings»-metoden, screenes sekvensen til det variable domenet av et gnagerantistoff mot hele biblioteket av kjente humane sekvenser for variable domener. Den humane sekvensen som er nærmest sekvensen til gnageren blir så akseptert som den humane rammen (FR) for det humaniserte antistoffet. Sims et al., J. Immunol., 11:2296 (1993); Chothia et al., J. Mol. Biol., 196:901 (1987). En annen metode er å anvende et spesielt ramme avledet fra konsensussekvensen til alle humane antistoffer av en spesiell undergruppe av lette eller tunge kjeder. Den samme rammen kan anvendes for flere forskjellige humaniserte antistoffer. Carter et al., Proc. Natl. Acad. Sci. USA, 89:428 (1992); Presta et al., J. Immunol., 11:2623 (1993) Det er dessuten viktig at antistoffer beholder høy affinitet for antigenet og andre fordelaktige biologiske egenskaper når de humaniseres. For å oppnå dette målet, fremstilles humaniserte antistoffer ifølge en foretrukket fremgangsmåte ved en prosess for analyse av opphavssekvensene og forskjellige konseptuelle humaniserte produkter ved hjelp av tredimensjonale modeller av de opphavssekvensene og de humaniserte sekvensene. Tredimensjonale immunoglobulinmodeller er vanligvis tilgjengelige og kjente for fagfolk. Det er tilgjengelig dataprogrammer som illustrerer og viser sannsynlige tredimensjonale konformasjonsstrukturer av valgte kandidatimmunoglobulinsekvenser. Inspisering av disse visningene gjør det mulig å analysere den sannsynlige rollen til restene i funksjonen til kandidatimmunoglobulinsekvensen, dvs. analyse av rester som påvirker immunoglobulinkandidatens evne til å binde antigenet sitt. På denne måten kan FR-rester velges og kombineres fra mottaker- og importsekvensene slik at man oppnår den ønskede antistoffegenskapen, som f.eks. økt affinitet for målantigenet/målantigenene. Generelt er CDR-restene direkte og mest vesentlig involvert i å påvirke antigenbindingen. Det er påtenkt forskjellige former for det humaniserte antistoffet. For eksempel kan det humaniserte antistoffet være et antistoffragment, som f.eks. et Fab, som eventuelt konjugeres med ett eller flere cytotoksiske midler for å lage et immunkonjugat. Alternativt kan det humaniserte antistoffet være et intakt antistoff, som f.eks. et intakt IgG1-antistoff. 4) Humane antistoffer
84 82 Som et alternativ til humanisering kan det lages humane antistoffer. For eksempel er det nå mulig å lage transgene dyr (for eksempel mus) som ved immunisering kan produsere et fullstendig repertoar av humane antistoffer uten endogen immunoglobulinproduksjon. For eksempel har det blitt beskrevet at den homozygote slettingen av genet for sammenføyingsregionen til den tunge kjeden av antistoffet (J H ) i kimære og kimbanemutante mus fører til fullstendig hemming av den endogene antistoffproduksjonen. Overføring av immunoglobulingenmatrisen til den humane kimbanen i slike kimbanemutantmus vil føre til at det produseres humane antistoffer ved antigenutfordring. Se f.eks. Jakobovits et al., Proc. Natl. Acad. Sci. USA, 90:21 (1993); Jakobovits et al., Nature, 362:2 28 (1993); Bruggermann et al., Year in Immuno., 7:33 (1993); U.S. patent nr og WO 97/ Alternativt kan bakteriofagdisplayteknologi anvendes til å produsere humane antistoffer og antistoffragmenter in vitro, fra genrepertoarer for variable immunoglobulindomener (V) fra ikke-immuniserte donorer. McCafferty et al., Nature 348:2 3 (1990); Hoogenboom and Winter, J. Mol. Biol. 227: 381 (1991). Ifølge denne metoden klones antistoffets V-domenegener i rammen til enten et stort eller lite kappeproteingen fra en trådformet bakteriofag, som f.eks. M13-eller fd, og vises som funksjonelle antistoffragmenter på overflaten til bakteriofagpartikkelen. Ettersom den trådformede partikkelen inneholder en enkelttrådet DNA-kopi av bakteriofaggenomet, fører valg ut fra de funksjonelle egenskapene til antistoffet også til valg av genet som koder for antistoffet som oppviser disse egenskapene. Dermed etterligner bakteriofagen noen av egenskapene til B-cellen. Bakteriofagdisplayet kan utføres i en rekke formater, gjennomgått i for eksempel i Johnson, Kevin S. og Chiswell, David J., Curr. Opin Struct. Biol. 3:64 71 (1993). Flere kilder for V-gensegmentene kan anvendes til bakteriofagdisplay. Clackson et al., Nature 32: (1991) isolerte en variert utvalg av antistoffer mot oksazolon fra et lite tilfeldig kombinatorisk bibliotek av V- gener fra milten til immuniserte mus. Et repertoar av V-gener fra uimmuniserte humane donorer kan konstrueres, og antistoffer mot et variert utvalg av antigener (inkludert selvantigener) kan isoleres stort sett ved å følge metodene som er beskrevet av Marks et al., J. Mol. Biol. 222:81 97 (1991) eller Griffith et al., EMBO J. 12: (1993). Se også U.S. patent nr og
85 83 1 Metodene til Cole et al. og Boerner et al. er også tilgjengelig for fremstillingen av humane monoklonale antistoffer (Cole et al., Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, s. 77 (198) og Boerner et al., J. Immunol. 147(1): 86 9 (1991). På samme måte kan humane antistoffer fremstilles ved å innføre humane immunoglobulinlokuser i transgene dyr, for eksempel mus der de endogene immunoglobulingenene er delvis eller fullstendig inaktivert. Ved utfordring observeres det human antistoffproduksjon, som på alle måter ligner veldig det man ser hos mennesker, inkludert genomorganiseringen, monteringen og antistoffrepertoaret. Denne tilnærmingen er beskrevet f.eks. i U.S. patent nr ; 4 806, 69 82, , , og i de følgende vitenskapelige publikasjonene: Marks et al., Bio/Technology : (1992); Lonberg et al., Nature 368: (1994); Morrison, Nature 368: (1994), Fishwild et al., Nature Biotechnology 14: 84 1 (1996), Neuberger, Nature Biotechnology 14: 826 (1996) og Lonberg og Huszar, Intern. Rev. Immunol. 13: 6 93 (199). Endelig kan humane antistoffer også lages in vitro med aktiverte B-celler (se U.S. patent nr og ). 20 () Antistoffragmenter Under visse omstendigheter er der fordeler ved å anvende antistoffragmenter i stedet for hele antistoffer. Mindre fragmentstørrelser gir rask utskilling, og kan gi forbedret tilgang til faste tumorer. Det er utviklet forskjellige metoder for fremstilling av antistoffragmenter. Tradisjonelt ble disse fragmentene avledet ved proteolytisk digerering av intakte antistoffer (se f.eks. Morimoto et al., J Biochem Biophys. Method. 24:7 117 (1992); og Brennan et al., Science 229:81 (198)). I dag kan disse fragmentene imidlertid fremstilles direkte av rekombinante vertsceller. Fab-, Fv- og scfvantistoffragmenter kan alle uttrykkes i og skilles ut fra E. coli, noe gjør det enkelt å fremstille store mengder av disse fragmentene. Antistoffragmenter kan isoleres fra antistofbakteriofagbibliotekene som er omtalt ovenfor. Alternativt kan Fab'-SH-fragmentene utvinnes direkte fra E. coli og bindes kjemisk for å danne F(ab') 2 -fragmenter (Carter et al., Bio/Technology : (1992)). Ifølge en annen tilnærming kan F(ab') 2 -fragmenter isoleres direkte fra rekombinant vertscellekultur. Fab og F(ab') 2 med økt halveringstid in vivo er
86 84 beskrevet i U.S. patent nr I andre utførelsesformer er det valgte antistoffet et enkeltkjedet Fv-fragment (scfv). Se WO 93/1618; U.S. patent nr og U.S. patent nr Antistoffragmentet kan også være et «lineært antistoff», som f.eks. beskrevet i U.S. patent Slike lineære antistoffragmenter kan være monospesifikke eller bispesifikke. 6) Antistoffavhengig enzymformidlet prodrugbehandling (ADEPT) Antistoffene ifølge den foreliggende oppfinnelsen kan også anvendes i ADEPT ved å konjugere antistoffet til et prodrugaktiverende enzym som omdanner en prodrug (for eksempel et kjemoterapeutisk peptidylmiddel, se WO 81/0114) til et aktivt legemiddel mot kreft. Se for eksempel WO 88/07378 og U. S. patent nr Enzymbestanddelen av immunkonjugatet som kan anvendes til ADEPT, inkluderer ethvert enzym som kan virke slik på en prodrug at den omdannes til den mer aktive cytotoksiske formen Enzymer som kan anvendes i fremgangsmåten ifølge denne oppfinnelsen inkluderer, men er ikke begrenset til, glykosidase, glukoseoksidase, humant lysozym, human glukuronidase, alkalisk fosfatase som kan anvendes til å omdanne fosfatholdige prodruger til frie legemidler; arylsulfatase som kan anvendes til å omdanne sulfatholdige prodruger til frie legemidler; cytosindeaminase som kan anvendes til å omdanne ikke-toksisk -fluorcytosin til kreftlegemidlet -fluoruracil; proteaser, som f.eks. serratiaprotease, termolysin, subtilisin, karboksypeptidaser (f.eks. karboksypeptidase G2 og karboksypeptidase A) og katepsiner 30 3 (som f.eks. katepsin B og L), som kan anvendes til å omdanne peptidholdige prodruger til frie legemidler; D-alanylkarboksypeptidaser, som kan anvendes til å omdanne prodruger som inneholder D-aminosyresubstituenter; karbohydratspaltende enzymer som f.eks. β-galaktosidase og nevraminidase som kan anvendes til å omdanne glykosylerte prodruger til frie legemidler; β- laktamase som kan anvendes til å omdanne legemidler derivatisert med β- laktamer til frie legemidler; og penicillinamidaser, som for eksempel penicillin V- amidase eller penicillin G-amidase, som kan anvendes til å omdanne legemidler derivatisert på aminnitrogenatomene med henholdsvis fenoksyacetyl- eller fenylacetylgrupper, til frie legemidler. Alternativt kan antistoffer med enzymatisk aktivitet, også kjent i faget som «abzymer» anvendes til å omdanne prodruger
87 8 ifølge oppfinnelsen til frie aktive legemidler (se f.eks. Massey, Nature 328: (1987)). Antistoff/abzym-konjugater kan fremstilles som beskrevet i dette dokumentet for framføring av abzymet til en tumorcellepopulasjon. De ovennevnte enzymene kan bindes kovalent til polypeptidet eller antistoffer som er beskrevet i dette dokumentet ved metoder godt kjent i faget, for eksempel ved å anvende de heterobifunksjonelle kryssbindingsmidlene som er omtalt ovenfor. Alternativt kan fusjonsproteiner som omfatter minst den antigenbindende regionen av antistoffet ifølge oppfinnelsen bundet til minst én funksjonelt aktiv del av et enzym ifølge oppfinnelsen konstrueres ved å anvende rekombinant DNA-metoder som er godt kjent i faget (se f.eks. Neuberger et al., Nature 312: (1984)). 7) Bispesifikke og polyspesifikke antistoffer 1 20 Bispesifikke antistoffer (BsAb) er antistoffer som har bindingsspesifisitet for minst to forskjellige epitoper, inkludert epitoper på det samme proteinet eller et annet protein. Alternativt kan en arm armeres for å binde til målantigenet, og en annen arm kan kombineres med en arm som binder til et utløsende molekyl på en leukocytt, som for eksempel et T-cellereseptormolekyl (f.eks. CD3), eller Fcreseptorer for IgG (FcγR) som f.eks. FcγR1 (CD64), FcγRII (CD32) og FcγRIII (CD 16), for å fokusere og lokalisere cellulære forsvarsmekanismer mot den målantigenuttrykkende cellen. Slike antistoffer kan avledes fra fullengdeantistoffer eller antistoffragmenter (for eksempel F(ab') 2 -bispesifikke antistoffer) Bispesifikke antistoffer kan også anvendes til å lokalisere cytotoksiske midler til celler som uttrykker målantigenet. Slike antistoffer har en arm som binder det ønskede antigenet og en annen arm som binder det cytotoksiske midlet (f.eks. saporin, antiinterferon a, vinkaalkoloid, ricin A-kjede, metotreksat eller radioisotophapten). Eksempler på kjente bispesifikke antistoffer inkluderer anti-erbb2/anti-fcgriii (WO 96/16673), anti-erbb2/anti-fcgri (U.S.P ), anti-erbb2/anti-cd3 (U.S.P ). 3 Fremgangsmåter for å fremstille bispesifikke antistoffer er kjent i faget. Tradisjonell produksjon av bispesifikke fullengdeantistoffer bygger på kouttrykking av to tung/lett immunoglobinkjedepar, der de to kjedene har forskjellig spesifisitet. Millstein et al., Nature, 30:37 39 (1983). På grunn av det tilfeldige utvalget av tunge og lette immunoglobulinkjeder produserer disse hybridomene
88 86 (kvadromer) en potensiell blanding av forskjellige antistoffmolekyler, men bare ett av dem har den korrekte bispesifikke strukturen. Rensingen av det korrekte molekylet, som vanligvis gjøres ved affinitetskromatografitrinn, er temmelig tungvinn, og produktutbyttene er lave. Lignende prosedyrer er offentliggjort i WO 93/08829 og i Traunecker et al., EMBO J., : (1991) Ifølge en annen tilnærming fusjoneres variable antistoffdomener med de ønskede bindingsspesifisitetene (antistoff-antigenkombinerende seter) til immunoglobulinsekvenser for konstante domene. Fusjonen gjøres fortrinnsvis med et konstant domene i den tunge immunoglobulinkjeden, som i det minste omfatter en del av hengsel-, CH2- og CH3-regionene. Det foretrekkes å ha den første konstante tungkjederegionen (CH1) som inneholder setet som er nødvendig for å binde den litte kjeden til stede i minst én av fusjonene. DNA som koder for de tunge immunoglobulinkjedefusjonene, og, hvis ønskelig, den lette immunoglobulinkjeden, settes inn i separate uttrykkingsvektorer, og kotransfekteres inn i en egnet vertsorganisme. Dette gir stor fleksibilitet i justeringen av de gjensidige proporsjonene mellom de tre polypeptidfragmentene i utførelsesformer når ulike forhold av de tre polypeptidkjedene som anvendes i konstruksjonen, gir det optimale utbyttet. Det er imidlertid mulig å sette inn kodingssekvensene for to av eller alle de tre polypeptidkjedene i én og samme uttrykkingsvektor når uttrykkingen av minst to polypeptidkjeder i like store forhold fører til høyt utbytte, eller når forholdene er av uvesentlig betydning I en foretrukket utførelsesform av denne tilnærmingen er de bispesifikke antistoffene sammensatt av en hybrid tung immunoglobulinkjede med en første bindingsspesifisitet i en arm og et hybridisert tungt/lett immunoglobulinkjedepar (som gir en andre bindingsspesifisitet) i den andre armen. Det ble funnet at denne asymmetriske strukturen letter separeringen av den ønskede bispesifikke forbindelsen fra uønskede immunoglobulinkjedekombinasjoner, siden nærværet av en lett immunoglobulinkjede i bare en halvdel av de bispesifikke molekylene gir en enkel separasjonsmåte. Denne tilnærmingen er offentliggjort i WO 94/ Flere opplysninger om produksjon av bispesifikke antistoffer finnes for eksempel Suresh et al., Methods in Enzymology 121: 2 (1986). 3 Ifølge en annen tilnærming som beskrives i WO 96/27011 eller U.S.P , kan grenseflaten mellom et par av antistoffmolekyler konstrueres for å maksimere prosentandelen av heterodimerer som utvinnes fra en rekombinant cellekultur. Den
89 87 foretrukne grenseflaten omfatter minst en del av CH3-regionen til et konstant antistoffdomene. I denne fremgangsmåten byttes én eller flere små aminosyresidekjeder fra grenseflaten til det første antistoffmolekylet ut med større sidekjeder (f.eks. tyrosin eller tryptofan). Kompenserende «hulrom» med identisk eller tilsvarende størrelse som de(n) store sidekjeden(e) lages på grenseflaten til det andre antistoffmolekylet ved å erstatte store aminosyresidekjeder med mindre (f.eks. alanin eller treonin). Dette gir en mekanisme for å øke utbyttet av heterodimeren i forhold til andre uønskede sluttprodukter som f.eks. homodimerer Metoder for å lage bispesifikke antistoffer av antistoffragmenter er beskrevet i litteraturen. For eksempel kan bispesifikke antistoffer fremstilles ved kjemisk binding. Brennan et al., Science 229: 81 (198) beskriver en prosedyre der intakte antistoffer spaltes proteolytisk for å lage F(ab') 2 -fragmenter. Disse fragmentene reduseres i nærvær av ditiolkompleksdanneren natriumarsenitt for å stabilisere nærliggende ditioler og hindre intermolekylær disulfiddanning. Fab'- fragmentene som dannes blir så omdannet til tionitrobenzoatderivater (TNB). Ett av Fab'-TNB-derivatene omdannes deretter tilbake til Fab'-TNB-derivatet for å danne det bispesifikke antistoffet. De bispesifikke antistoffene som produseres kan anvendes som midler for selektiv immobilisering av enzymer. Fab'-fragmentene kan utvinnes direkte fra E. coli og bindes sammen kjemisk til bispesifikke antistoffer. Shalaby et al., J. Exp. Med. 17: (1992) beskriver produksjonen av fullt humaniserte bispesifikke antistoff-f(ab') 2 - molekyler. Hvert Fab'-fragment ble separat utskilt fra E. coli og utsatt for retningsbestemt kjemisk sammenbinding in vitro for å danne det bispesifikke antistoffet. Det bispesifikke antistoffet som ble dannet på denne måten var i stand til å binde til celler som overuttrykker ErbB2-reseptoren og normale humane T-celler, samt at det utløste den lytiske aktiviteten til humane cytotoksiske lymfocytter mot humane brysttumormål. Det er også beskrevet forskjellige metoder for fremstilling og isolering av toverdige antistoffragmenter direkte fra rekombinant cellekultur. For eksempel har toverdige heterodimerer blitt produsert ved å anvende leucinglidelåser. Kostelny et al., J. Immunol., 148(): (1992). Leucinglidelåspeptidene fra Fos- og Jun-proteinene ble bundet til Fab'-delene av to forskjellige antistoffer ved genfusjon. Antistoffhomodimerene ble redusert i hengselregionen for å danne monomerer og deretter oksydert igjen for å
90 88 danne antistoffheterodimerene. «Dialegeme»-teknologien, beskrevet av Hollinger et al., Proc. Natl. Acad. Sci. USA, 90: (1993) har gitt oss en alternativ mekanisme for å lage bispesifikke/toverdige antistoffragmenter. Fragmentene omfatter et variabelt tungkjededomene (V H ) koblet til et variabelt lettkjededomene (V L ) av et bindeledd som er for kort til å tillate paring mellom de to domenene på den samme kjeden. Følgelig tvinges V H - og V L -domenene i et fragment til å pares med de komplementære V L - og V H -domenene i et annet fragment, og dermed danne to antigenbindingsseter. Det er også rapportert en annen strategi for å lage bispesifikke/toverdige antistoffragmenter ved hjelp av enkeltkjede-fv-(sfv)-dimerer. Se Gruber et al., J. Immunol., 12:368 (1994). Antistoffer med mer enn to valenser er påtenkt. Det kan f.eks. fremstilles trispesifikke antistoffer. Tutt et al., J. Immunol. 147: 60 (1991) Eksempler på bispesifikke antistoffer kan binde to forskjellige epitoper på et gitt molekyl. Alternativt kan en antiproteinarm kombineres med en arm som binder et utløsende molekyl på en leukocytt, som for eksempel et T-cellereseptormolekyl (f.eks. CD2, CD3, CD28 eller B7), eller Fc-reseptorer for IgG (FcγR), som f.eks. FcγRI (CD64), FcγRII (CD32) og FcγRIII (CD 16), for å fokusere celleforsvarsmekanismer til cellen som uttrykker det bestemte proteinet. Bispesifikke antistoffer kan også anvendes til å lokalisere cytotoksiske midler til celler som uttrykker et bestemt protein. Slike antistoffer har en proteinbindingsarm og en arm som binder et cytotoksisk middel eller en radionuklidekelatdanner, som f.eks. EOTUBE, DPTA, DOTA eller TETA. Et annet bispesifikt antistoff av interesse binder proteinet av interesse, og binder også vevsfaktor (TF). 8) Flerverdige antistoffer 30 3 En celle som uttrykker et antigen som antistoffene binder til, kan raskere internalisere (og/eller katabolisere) et flerverdig antistoff enn et toverdig antistoff. Antistoffene ifølge den foreliggende oppfinnelsen kan være flerverdige antistoffer (som er forskjellige fra IgM-klassen) med tre eller flere antigenbindingsseter (f.eks. fireverdige antistoffer), som lett kan fremstilles ved rekombinant uttrykking av nukleinsyre som koder for polypeptidkjedene i antistoffet. Det flerverdige antistoffet kan omfatte et dimeriseringsdomene og tre eller flere antigenbindingsseter. Det foretrukne dimeriseringsdomenet omfatter (eller består av) en Fc-region eller en hengselregion. I dette scenariet vil antistoffet omfatte en
91 89 1 Fc-region og tre eller flere antigenbindingsseter nær aminoenden av Fc-regionen. Det foretrukne flerverdige antistoffet i dette dokumentet omfatter (eller består av) tre til rundt åtte, men fortrinnsvis fire antigenbindingsseter. Det flerverdige antistoffet omfatter minst én polypeptidkjede (og fortrinnsvis to polypeptidkjeder), der polypeptidkjeden(e) omfatter to eller flere variable domener. For eksempel kan polypeptidkjeden(e) omfatte VD1-(X1) n -VD2-(X2) n -Fc, der VD1 er et første variabelt domene, er VD2 er et andre variabelt domene, Fc er en polypeptidkjede i en Fc-region, X1 og X2 representerer en aminosyre eller polypeptid, og n er 0 eller 1. For eksempel kan polypeptidkjeden(e) omfatte: VH-CH1-fleksibelt bindeledd - VH-CH1-Fc-regionkjeden; eller VH-CH1-VH-CH1-Fc-regionkjeden. Det flerverdige antistoffet i dette dokumentet omfatter fortrinnsvis også minst to (og fortrinnsvis fire) polypeptider fra det variable domenet i den lette kjeden. Det flerverdige antistoffet i dette dokumentet kan for eksempel omfatte fra rundt to til rundt åtte polypeptider fra det variable domenet i den lette kjeden. Polypeptidene fra det variable domenet i den lette kjeden som er påtenkt i dette dokumentet omfatter et variabelt domene i den lette kjeden og omfatter eventuelt også et CL-domene. 9) Heterokonjugatantistoffer Heterokonjugatantistoffer er også innenfor omfanget til den foreliggende oppfinnelsen. Heterokonjugatantistoffer er sammensatt av to kovalent sammenbundne antistoffer. For eksempel kan ett av antistoffene i heterokonjugatet være bundet til avidin, og det andre til biotin. Slike antistoffer har for eksempel blitt foreslått for å målsøke immunsystemceller mot uønskede celler, U.S.P , og for å behandle HIV-infeksjon. WO 91/00360, WO 92/ og EP Det påtenkes at antistoffene også kan fremstilles in vitro ved kjente metoder innen syntetisk proteinkjemi, inkludert slike som involverer kryssbindingsmidler. For eksempel kan immuntoksiner konstrueres ved å anvende en disulfidutvekslingsreaksjon eller ved å danne en tioeterbinding. Eksempler på egnede reagenser for dette formålet inkluderer iminotiolat og metyl-4-merkaptobutyrimidat og slike som offentliggjøres f.eks. i U.S. patent nr Heterokonjugatantistoffer kan fremstilles ved å anvende hvilke som helst hensiktsmessige kryssbindingsmetoder. Egnede kryssbindingsmidler er godt kjent i faget, og er offentliggjort i US patent nr , sammen med flere kryssbindingsmetoder. ) Konstruering av effektorfunksjonen
92 90 1 Det kan være ønskelig å modifisere antistoffet ifølge oppfinnelsen når det gjelder effektorfunksjon, for eksempel for å forbedre den antigenavhengige celleformidlede cytotoksisiteten (ADCC) og/eller den komplementavhengige cytotoksisiteten (CDC) til antistoffet. Dette kan oppnås ved å innføre én eller flere aminosyreerstatninger i en Fc-region av antistoffet. Alternativt eller i tillegg kan (en) cysteinrest(er) innføres i Fc-regionen slik at det kan dannes disulfidbindinger mellom kjedene i denne regionen. Det homodimere antistoffet som dannes på denne måten kan ha forbedret internaliseringsevne og/eller økt komplementformidlet celledreping og antistoffavhengig cellecytotoksisitet (ADCC). Se Caron et al., J. Exp Med. 176: (1992) og Shopes, B. J. Immunol. 148: (1992). Homodimere antistoffer med økt antitumoraktivitet kan også fremstilles ved å anvende heterobifunksjonelle kryssbindere som beskrevet i Wolff et al., Cancer Research 3: (1993). Alternativt kan det konstrueres et antistoff som har doble Fc-regioner, og dermed kan ha økte komplementlyserings- og ADCC-evner. Se Stevenson et al., Anti-Cancer Drug Design 3: (1989). 20 For å øke serumhalveringstiden til antistoffet, kan man føre en stabiliserende («salvage») reseptorbindingsepitop inn i antistoffet (spesielt et antistoffragment) for eksempel som beskrevet i U.S. patent Slik det anvendes i dette dokumentet står uttrykket «stabiliserende reseptorbindingsepitop» for en epitop i Fc-regionen til et IgG-molekyl (f.eks. IgG 1, IgG 2, IgG 3 eller IgG 4 ) som er ansvarlig for å øke serumhalveringstiden til IgG-molekylet in vivo. 2 11) Immunkonjugater 30 Oppfinnelsen vedrører også immunkonjugater eller antistoff/legemiddel-konjugater (ADC), som omfatter et antistoff konjugert til et cytotoksisk middel som f.eks. et kjemoterapeutisk middel, toksin (f.eks. et enzymatisk aktivt toksin av bakterie-, sopp-, plante- eller dyreopprinnelse, eller fragmenter av disse), eller en radioaktiv isotop (dvs. et radiokonjugat). En slik ADC må vise akseptabel sikkerhetsprofil. 3 Anvendelsen av ADC til lokal framføring av cytotoksiske eller cytostatiske midler, for eksempel legemidler for å drepe eller hemme tumorceller under behandling av kreft [Syrigos and Epenetos, Anticancer Research 19: (1999); Niculeascu-Duvaz and Springer, Adv. Drug Del. Rev. 26: (1997); US ] gjøt det teoretisk mulig å målrette levering av legemiddelgruppen til tumorer, og
93 intracellulær opphoping i denne, der systemisk administrering av disse ukonjugerte legemiddelmidlene kan føre til uakseptable toksisitetskonsentrasjoner både overfor normale celler, og overfor tumorceller som søkes fjernet (Baldwin et al., Lancet, (1986); Thorpe, (198) Antibody Carriers Of Cytotoxic Agents In Cancer Therapy: En gjennomgang, i Monoclonal Antibodies '84: Biological And Clinical Applications, A. Pinchera et al. (red.), s ). Derfor søker man maksimal virkning med minimal toksisitet. Både polyklonale antistoffer og monoklonale antistoffer er rapportert som nyttige i disse strategiene (Rowland et al., Cancer Immunol. Immunother. 21: (1986)). Legemidlene som anvendes i disse metodene inkluderer daunomycin, doksorubicin, metotreksat og vindesin. Toksiner som anvendes i antistofftoksinkonjugater inkluderer bakterielle toksiner som difteritoksin, plantetoksiner som f.eks. ricin, toksiner med små molekyler, som f.eks. geldanamycin (Mandler et al. J. Nat. Cancer Inst. 92(19): (2000); Mandler et al., Bioorganic & Med. Chem. Letters :2 28 (2000); Mandler et al. Bioconjugate Chem. 13: (2002)), maytansinoider (EP ; Liu et al., Proc. Natl. Acad. Sci. USA 93: (1996)), og kalikeamicin (Lode et al., Cancer Res. 8:2928 (1998); og Hinman et al., Cancer Res. 3: (1993)). Toksinene kan utøve sine cytotoksiske og cytostatiske virkninger ved mekanismer som inkluderer tubulinbinding, DNA-binding eller topoisomerasehemming. Noen cytotoksiske legemidler er tilbøyelige til å være inaktive eller mindre aktive når de konjugeres til store antistoffer eller proteinreseptorligander. 2 Kjemoterapeutiske midler som er nyttige til å lage slike immunkonjugater beskrives ovenfor, og inkluderer BCNU, streptozoicin, vinkristin, vinblastin, adriamycin og -fluoruracil Enzymatisk aktive toksiner og fragmenter av disse som kan anvendes inkluderer difteri A-kjeden, ikke-bindende aktive fragmenter av difteritoksin, eksotoksin A- kjeden (fra Pseudomonas aeruginosa), ricin A-kjeden, abrin-a-kjeden, modeccin-akjeden, alfa-sarcin, Aleurites fordii-proteiner, diantinproteiner, Phytolaca americanaproteiner (PAPI, PAPII og PAP-S), Momordica charantia-hemmer, kurcin, krotin, saponaria officinalis-hemmer, gelonin, mitogellin, restriktosin, fenomycin, enomycin, og trikotecenene. En rekke radionuklider er tilgjengelige for produksjon av radiokonjugerte antistoffer. Eksempler inkluderer 212 Bi, 131 I, 131 In, 90 Y og 186 Re. Konjugater av antistoffet og det cytotoksiske midlet lages ved å anvende flere bifunksjonelle proteinbindingsmidler som f.eks. N-suksinimidyl-3-(2- pyridylditiol)propionat (SPDP), iminotiolan (IT), bifunksjonelle derivater av
94 imidoestere (som f.eks. dimetyladipimidat-hcl), aktive estere (som f.eks. disuksinimidylsuberat), aldehyder (som f.eks. glutaraldehyd), bisazidoforbindelser (som f.eks. bis-(p-azidobenzoyl)heksandiamin), bisdiazoniumderivater (som f.eks. bis-(p-diazoniumbenzoyl)-etylendiamin), diisocyanater (som f.eks. toluen-2,6- diisocyanat) og bis-aktive fluorforbindelser (som f.eks. 1,-difluor-2,4- dinitrobenzen). For eksempel kan et ricinimmuntoksin fremstilles som beskrevet i Vitetta et al., Science, 238: 98 (1987). Karbon-14-merket 1-isotiocyanatobenzyl- 3-metyldietylentriaminpentaeddiksyre (MX-DTPA) er et eksempel på en kelatdanner for konjugering av radionukleotid til antistoffet. Se f.eks. WO 1994/126. Konjugatene av antistoffet og det cytotoksiske midlet lages ved å anvende flere bifunksjonelle proteinbindingsmidler som f.eks. N-suksinimidyl-3-(2- pyridylditiol)propionat (SPDP), iminotiolan (IT), bifunksjonelle derivater av imidoestere (som f.eks. dimetyladipimidat HCl), aktive estere (som f.eks. disuksinimidylsuberat), aldehyder (som f.eks. glutaraldehyd), bisazidoforbindelser (som f.eks. bis-(p-azidobenzoyl)heksandiamin), bisdiazoniumderivater (som f.eks. bis-(p-diazoniumbenzoyl)-etylendiamin), diisocyanater (som f.eks. toluen-2,6-diisocyanat) og bis-aktive fluorforbindelser (som f.eks. 1,-difluor-2,4-dinitrobenzen). Et ricinimmuntoksin fremstilles f.eks. som beskrevet i Vitetta et al., Science, 238: 98 (1987). Karbon-14-merket 1- isotiocyanatobenzyl-3-metyldietylentriaminpentaeddiksyre (MX-DTPA) er et eksempel på en kelatdanner for konjugering av radionukleotid til antistoffet. Se WO94/126. Bindeleddet kan være et «spaltbart bindeledd» som letter frigjøring av det cytotoksiske legemidlet i cellen. Det kan f.eks. anvendes et syrelabilt bindeledd, peptidasefølsomt bindeledd, dimetylbindeledd eller disulfidholdig bindeledd (Chari et al., Cancer Res. 2: (1992)) I tillegg er de små molekyltoksinene som f.eks. kalikeamicin, maytansin (U.S.P ), trikoten og CC6 også påtenkt som konjugerbare toksiner som kan anvendes til den nye formuleringen. I en utførelsesform kan antistoffet i sin fulle lengde eller antigenbindende fragmenter av dette konjugeres til ett eller flere maytansinoidmolekyler (for eksempel rundt 1 til rundt maytansinoidmolekyler per antistoffmolekyl). Maytansinoider er mitotiske hemmere som virker ved å hemme tubulinpolymeriseringen. Maytansinoider, isolert fra naturlige kilder eller fremstilt syntetisk, inkludert maytansin, maytansinal og derivater og analoger av disse er blitt beskrevet, se f.eks. U.S. patent nr og referanser som siteres der (se kol. 2, linje 3 til kol. 3, linje ) og U.S. patent og 4
95 Fremgangsmåter for fremstilling av antistoff/maytansinoid-konjugater er også beskrevet i U.S. pat. nr I en foretrukket utførelsesform bindes et maytansinoid til antistoffet gjennom en disulfidgruppen eller en annen svovelholdig bindeleddgruppe. Maytansin kan for eksempel omdannes til May-SS- Me, som kan reduseres til May-SH3 og omsettes med modifisert antistoff til et maytansinoid/antistoff-immunkonjugat. Chari et al., Cancer Res. 2: (1992). Antistoffet kan modifiseres ved kjente fremgangsmåter, og antistoffet som inneholder frie eller beskyttede tiolgrupper omsettes deretter med et disulfidholdig maytansinoid for å fremstille konjugatet. Cytotoksisiteten til antistoff/maytansinoid-konjugatet kan måles in vitro eller in vivo ved hjelp av kjente fremgangsmåter og den bestemte IC Kalikeamicin er et annet immunkonjugat av interesse. Kalikeamicinfamilien av antibiotika er i stand til å produsere dobbelttrådede DNA-pauser ved under pikomolare konsentrasjoner. Strukturelle analoger av kalikeamicin som kan anvendes inkluderer, men er ikke begrenset til, γ 1 1, α 1 2, α 1 3, N-aceytl-γ 1 1, PSAG og θ 1 1 (Hinman et al., Cancer Res. 3: (1993) og Lode et al., Cancer Res. 8: (1998)). Andre antitumorlegemidler antistoffet kan konjugeres til, inkluderer QFA, som er et antifolat. Både kalikeamicin og QFA har intracellulære virkningsseter og trenger ikke lett gjennom plasmamembranen. Derfor vil cellulært opptak av disse midlene gjennom antistofformidlet internalisering i høy grad øke de cytotoksiske virkningene deres. 2 Immunkonjugater dannet mellom et antistoff og en forbindelse med nukleolytisk aktivitet (for eksempel en ribonuklease eller DNA-endonuklease, som f.eks. deoksyribonuklease, DNase) er også påtenkt I ADC ifølge oppfinnelsen konjugeres et antistoff (Ab) til én eller flere legemiddeldeler (D), for eksempel rundt 1 til rundt 20 legemiddeldeler per antistoff, gjennom et bindeledd (L). ADC av formel I kan fremstilles ved forskjellige reaksjonsveier, der det benyttes organiske kjemiske reaksjoner, forhold og reagenser som er kjent for fagfolk, inkludert: (1) reaksjon mellom en nukleofil gruppe av et antistoff og en toverdig bindeleddreagens til Ab-L, gjennom en kovalent binding, og så reaksjon med en legemiddelgruppe D; og (2) reaksjon mellom en nukleofil gruppe med en legemiddelgruppe med en toverdig bindeleddreagens til D-L, gjennom en kovalent binding, og så reaksjon med den nukleofile gruppen i et antistoff.
96 94 1 Nukleofile grupper i antistoffer inkluderer, men er ikke begrenset til: (i) amingrupper i N-endene, (ii) amingrupper i sidekjedene, for eksempel lysin, (iii) tiolgrupper i sidekjedene, for eksempel cystein og (iv) sukkerhydroksyl- eller aminogrupper der antistoffet glykosyleres. Amin-, tiol- og hydroksylgrupper er nukleofile og kan reagere ved å danne kovalente bindinger med elektrofile grupper på bindeleddgrupper og bindeleddreagenser, inkludert: (i) aktive estere, som f.eks. NHS-estere, HOBt-estere, halogenformiater og syrehalogenider; (ii) alkyl- og benzylhalogenider som f.eks. halogenacetamider; og (iii) aldehyder, ketoner, karboksyl- og maleimidgrupper. Enkelte antistoffer har reduserbare disulfider mellom kjedene, dvs. cysteinbroer. Antistoffer kan gjøres reaktive for konjugering med bindeleddreagenser ved å behandle dem med et reduksjonsmiddel, som f.eks. DTT (ditiotreitol). Hver cysteinbro vil dermed teoretisk sett danne to reaktive tiolnukleofiler. Andre nukleofile grupper kan innføres i antistoffer ved reaksjonen mellom lysiner med 2-iminotiolan (Traut-reagens) som omdanner et amin til en tiol ADC ifølge oppfinnelsen kan også fremstilles ved å modifisere antistoffet ved å innføre elektrofile grupper, som kan reagere med nukleofile substituenter på bindeleddreagensen eller legemidlet. Sukkerartene til glykosylerte antistoffer kan oksyderes, f.eks. med oksiderende perjodatreagenser, for å danne aldehydeller ketogrupper som kan reagere med amingruppen i bindeleddreagenser eller legemiddelgrupper. De resulterende iminschiffbasegruppene kan danne en stabil binding, eller kan reduseres for eksempel med borhydridreagenser for å danne stabile aminbindinger. I en utførelsesform kan reaksjon mellom karbohydratdelen av et glykosylert antistoff og enten galaktoseoksidase eller natriummetaperjodat gi karbonylgrupper (aldehyd og keton) i proteinet som kan reagere med passende grupper på legemidlet (Domen et al., J. Chromatog., : (1990)). I en annen utførelsesform kan proteiner som har en serinrest eller treoninrest i N-enden reagere med natriummetaperjodat, som gir et aldehyd i stedet for den første aminosyren (Geoghegan and Stroh, Bioconjugate Chem. 3: (1992); US ). Et slikt aldehyd kan reageres med en legemiddelgruppe eller bindeleddnukleofil. Nukleofile grupper på en legemiddelgruppe inkluderer likeledes, men er ikke begrenset til: amin-, tiol-, hydroksyl-, hydrazid-, oksim-, hydrazin-,
97 9 tiosemikarbazon-, hydrazin-, karboksylat- og arylhydrazidgrupper som kan reagere og danne kovalente bindinger med elektrofile grupper på bindeledsgrupper og bindeleddreagenser inkludert: (i) aktive estere, som f.eks. NHS-estere, HOBt-estere, halogenformiater og syrehalogenider; (ii) alkyl- og benzylhalogenider som f.eks. halogenacetamider; og (iii) aldehyder, ketoner, karboksyl- og maleimidgrupper. Alternativt kan det fremstilles et fusjonsprotein som omfatter antistoffet og det cytotoksiske midlet, for eksempel ved rekombinante metoder eller peptidsyntese. DNA-lengden kan omfatte respektive regioner som koder for de to delene til konjugatet enten inntil hverandre eller adskilt med en region som koder for et bindeleddspeptid som ikke ødelegger de ønskede egenskapene til konjugatet I nok en annen utførelsesform kan antistoffet konjugeres til en «reseptor» (som f.eks. streptavidin) for å utnyttes i forberedelser til målretting i tumorer, der antistoffreseptorkonjugater administreres til pasienten, så fjernes ubundet konjugat fra sirkulasjonen ved å anvende et utskillingsmiddel og deretter administreres en «ligand» (f.eks. avidin) som konjugeres til et cytotoksisk middel (for eksempel et radionukleotid). ADC i dette dokumentet fremstilles eventuelt med kryssbindingsreagenser: BMPS, EMCS, GMBS, HBVS, LC-SMCC, MBS, MPBH, SBAP, SIA, SIAB, SMCC, SMPB, SMPH, sulfo-emcs, sulfo-gmbs, sulfo-kmus, sulfo-mbs, sulfo-siab, sulfo-smcc og sulfo-smpb og SVSB (suksinimidyl-(4-vinylsulfon)benzoat), som er kommersielt tilgjengelige (f.eks. Pierce Biotechnology, Inc., Rockford, IL) Antistoffet kan også konjugeres til et sterkt radioaktivt atom. En rekke radionuklider er tilgjengelige for produksjon av radiokonjugerte antistoffer. Eksempler inkluderer At 211, Bi 212, I 131, In 131, Y 90, Re 186, Re 188, Sm 13, P 32 og Pb 212 og radioaktive isotoper av Lu. Når konjugatet anvendes til diagnose, kan det omfatte et radioaktivt atom for scintigrafiske studier, f.eks. Tc 99 eller I 123 eller en spinnmerking for magnetresonansavbilding (NMR) (også kalt MRI), som for eksempel jod 123, jod 131, indium 111, fluor 19, karbon 13, nitrogen 1, oksygen 17, gadolinium, mangan eller jern. Radioaktive eller andre etiketter kan innføres i konjugatet på kjente måter. For eksempel kan peptidet biosyntetiseres eller syntetiseres ved kjemisk
98 96 aminosyresyntese med egnede aminosyreforløpere som for eksempel har fluor 19 i stedet for hydrogen. Merkinger som f.eks. Tc 99 eller I 123 Re 186, Re 188 og In 111 kan festes med en cysteinrest i peptidet. Yttrium 90 kan festes med en lysinrest. IODOGEN -metoden kan anvendes til å innføre jod 123, Fraker et al., Biohem. Biophys. Res. Commun. 80:49 7 (1978). Andre metoder for konjugering av radionuklider er beskrevet i «Monoclonal Antibodies in Immunoscintigraphy», (Chatal, CRC Press 1989). Alternativt kan et fusjonsprotein som omfatter antistoffet og det cytotoksiske midlet fremstilles for eksempel ved rekombinante metoder eller peptidsyntese. DNA-lengden kan omfatte respektive regioner som koder for de to delene av konjugatet enten inntil hverandre eller adskilt med en region som koder for et bindeleddspeptid som ikke ødelegger de ønskede egenskapene til konjugatet I en annen utførelsesform kan antistoffet konjugeres til en «reseptor» (som f.eks. streptavidin) for å utnyttes i forberedelser til målretting i tumorer, der antistoffreseptorkonjugatet administreres til pasienten, så fjernes ubundet konjugat fra sirkulasjonen ved å anvende et utskillingsmiddel og deretter administreres en «ligand» (f.eks. avidin) som konjugeres til et cytotoksisk middel (for eksempel et radionukleotid). 12) Andre modifikasjoner av aminosyresekvensen Det påtenkes modifikasjon(er) av aminosyresekvensen til antistoffene som er beskrevet i dette dokumentet. For eksempel kan det være ønskelig å forbedre bindingsaffiniteten og/eller andre biologiske egenskaper ved antistoffet. Aminosyresekvensvarianter av antistoffet fremstilles ved å innføre passende nukleotidendringer til antistoffnukleinsyren, eller ved peptidsyntese. Slike modifikasjoner inkluderer for eksempel å fjerne og/eller sette inn og/eller bytte ut rester i aminosyresekvensene til antistoffet. En hvilken som helst kombinasjon av å fjerne, sette inn og bytte ut gjøres for å komme frem til den endelige konstruksjonen, forutsatt at den endelige konstruksjonen har de ønskede egenskapene. Aminosyreendringene kan også endre posttranslasjonelle prosesser i antistoffet, som f.eks. endring av antallet eller posisjonen til glykosyleringsseter. En nyttig metode for å identifisere visse rester eller regioner av antistoffet som er foretrukne seter for mutagenese kalles «alaninskanningsmutagenese» som
99 97 beskrevet av Cunningham and Wells i Science, 244:81 8 (1989). Her identifiseres en rest eller en gruppe av målrester (f.eks. ladde rester som f.eks. arg, asp, his, lys og glu) og byttes ut med en nøytral eller negativt ladet aminosyre (alanin eller polyalanin foretrekkes mest) for å påvirke interaksjonen mellom aminosyreantigenene. De aminosyreposisjonene som viser funksjonell følsomhet for erstatningene raffineres deretter ved å innføre flere eller andre varianter på eller for, erstatningsposisjonene. Posisjonen der det innføres en aminosyresekvensvariant er altså forhåndsbestemt, men mutasjonstypen i seg selv er ikke nødvendigvis forhåndsbestemt. For eksempel utføres ala-skanning eller tilfeldig mutagenese på målkodonet eller regionen for å analysere oppførselen til en mutasjon i en gitt posisjon, og de uttrykte antistoffvariantene screenes for den ønskede aktiviteten Innsettinger i aminosyresekvensen inkluderer både amino- og/eller karboksylendefusjoner som varierer i lengde fra en rest til polypeptider som inneholder hundre eller flere rester, og innsetting av en eller flere aminosyrerester inne i sekvensen. Eksempler på endeinnsetting inkluderer et antistoff med en metionylrest i N-enden eller antistoffet fusjonert til et cytotoksisk polypeptid. Andre innsettingsvarianter av antistoffmolekylet inkluderer fusjon mellom N- eller C-enden av antistoffet og et enzym (for eksempel for ADEPT) eller et polypeptid som øker serumhalveringstiden til antistoffet En annen varianttype er en aminosyresubstitueringsvariant. I disse variantene er minst én aminosyrerest i antistoffmolekylet substituert med en annen rest. Posisjonene som er av størst interesse for substitueringsmutagenese inkluderer de hypervariable regionene, men FR-endringer er også påtenkt. Konservative substitusjoner er vist i tabell A nedenfor under overskriften «foretrukne substitusjoner». Hvis slike substitusjoner fører til en endring i den biologiske aktiviteten, kan det innføres mer vesentlige endringer, betegnet «eksempelsubstitusjoner» i tabell A, eller som nærmere beskrevet nedenfor under henvisning til aminosyreklasser, og produktene screenes. TABELL A Aminosyresubstitusjoner Opprinneligrest Eksempelsubstitusjoner Foretrukne substitusjoner
100 98 Aminosyresubstitusjoner Opprinneligrest Eksempelsubstitusjoner Foretrukne substitusjoner Ala (A) val; leu; ile val Arg (R) lys; gln; asn lys Asn (N) gln; his; asp, lys; arg gln Asp (D) glu; asn glu Cys (C) ser; ala ser Gln (Q) asn; glu asn Glu (E) asp; gln asp Gly (G) ala ala His (H) asn; gln; lys; arg arg Ile (I) Leu (L) leu; val; met; ala; phe; norleucin leu norleucin; ile; val; met; ala; phe ile Lys (K) arg; gln; asn arg Met (M) leu; phe; ile leu Phe (F) leu; val; ile; ala; tyr tyr Pro (P) Ala ala Ser (S) Thr thr Thr (T) Ser ser Trp (W) tyr; phe tyr Tyr (Y) trp; phe; thr; ser phe Val (V) ile; leu; met; phe; ala; norleucin leu
101 99 Vesentlige endringer i de biologiske egenskapene til antistoffet oppnås ved å velge substitusjoner som avviker vesentlig i sin virkning på å opprettholde (a) strukturen til polypeptidryggraden i området for substitusjonen, for eksempel, som en flak- eller spiralformet konformasjon, (b) ladningen eller hydrofobisiteten til molekylet i målposisjonen, eller (c) omfanget av sidekjeden. Naturlig forekommende rester deles inn i grupper ut fra felles sidekjedeegenskaper: (1) hydrofobe: norleucin, met, ala, val, leu, ile; (2) nøytrale hydrofile: cys, ser, thr; (3) sure: asp, glu; (4) basiske: asn, gln, his, lys, arg; () rester som påvirker kjedeorienteringen: gly, pro; og (6) aromatiske: trp, tyr, phe. 1 Ikke-konservative substitusjoner vil medføre at et medlem av én av disse klassene byttes ut med et medlem av en annen klasse. 20 Enhver cysteinrest som ikke er involvert i å opprettholde den riktige konformasjonen til antistoffet kan også byttes ut, generelt med serin, for å forbedre den oksidative stabiliteten til molekylet og hindre avvikende kryssbinding. Omvendt kan antistoffet tilføres cysteinbinding(er) for å forbedre stabiliteten (særlig hvis antistoffet er et antistoffragment, som f.eks. et Fv-fragment) En spesielt foretrukket type substitusjonsvariant innebærer å bytte ut én eller flere rester fra den hypervariable regionen av et opphavsantistoff (for eksempel et humanisert eller humant antistoff). Vanligvis vil de(n) resulterende varianten(e) som er valgt for videreutvikling ha bedre biologiske egenskaper enn opphavsantistoffet som de lages av. En praktisk måte å lage slike substitusjonsvarianter på innebærer affinitetsmodning ved hjelp av bakteriofagdisplay. Kort fortalt muteres flere posisjoner i den hypervariable regionen (f.eks. 6 7 posisjoner) for å lage alle mulige aminosubstitusjoner i hver posisjon. Antistoffvariantene som lages på denne måten vises på en enverdig måte av trådformede bakteriofagpartikler som fusjoner til gen-iii-produktet av M13 som er pakket i hver partikkel. De bakteriofagviste variantene screenes deretter for biologiske aktivitet (for eksempel bindingsaffinitet) som beskrevet i dette dokumentet. For å identifisere kandidatposisjoner i den hypervariable regionen for modifisering, kan det uføres alaninskanningsmutagenese for å identifisere rester i den hypervariable regionen som bidrar signifikant til
102 0 antigenbindingen. Alternativt, eller i tillegg, kan det være fordelaktig å analysere krystallstrukturen til antigen/antistoff-komplekset for å identifisere kontaktpunktene mellom antistoffet og IgE. Slike kontaktrester og naborester er kandidater for substituering ifølge metodene som utdypes i dette dokumentet. Når det lages slike varianter, utsettes panelet av varianter for screening som beskrevet i dette dokumentet og antistoffer med overlegne egenskaper i én eller flere relevante analyser kan velges for videreutvikling. En annen type aminosyrevariant av antistoffet endrer det opprinnelige glykosyleringsmønsteret til antistoffet. Endre betyr her å slette én eller flere karbohydratgrupper som finnes i antistoffet og/eller tilsette ett eller flere glykosyleringsseter som ikke er til stede i antistoffet Glykosylering av antistoffer er vanligvis enten N-forbundet eller O-forbundet. N- binding betyr at karbohydratgruppen er festet til sidekjeden av en asparaginrest. Tripeptidsekvensene asparagin-x-serin og asparagin-x-treonin, der X er en hvilken som helst aminosyre bortsett fra prolin, er gjenkjennelsessekvensene for enzymatisk festing av karbohydratgruppen til asparaginsidekjeden. Altså danner nærværet av enhver av disse tripeptidsekvensene i et polypeptid et potensielt glykosyleringssete. O-bundet glykosylering betyr at én av sukkerartene N- acetylgalaktosamin, galaktose eller xylose festes til en hydroksyaminosyre, vanligvis serin eller treonin, med -hydroksyprolin eller -hydroksylysin kan også anvendes. 2 Det er praktisk å tilføre antistoffet glykosyleringsseter ved å endre aminosyresekvensen slik at den inneholder en eller flere av de ovenfor beskrevne tripeptidsekvensene (for N-bundne glykosyleringsseter). Endringen kan også gjøres ved å tilføye eller bytte ut én eller flere serin- eller treoninrester i sekvensen av det opprinnelige antistoffet (for O-bundne glykosyleringsseter) Nukleinsyremolekyler som koder for varianter av aminosyresekvensen til antistoffet mot IgE fremstilles ved en rekke metoder som er kjent i faget. Disse metodene inkluderer, men er ikke begrenset til, isolering fra en naturlig kilde (i tilfellet med naturlig forekommende aminosyresekvensvarianter) eller fremstilling ved oligonukleotidformidlet (eller seterettet) mutagenese, PCRmutagenese, og kassettmutagenese av en tidligere fremstilt variant eller en ikke-variantversjon av antistoffet mot IgE.
103 1 13) Andre antistoffmodifikasjoner Antistoffene ifølge den foreliggende oppfinnelsen kan modifiseres ytterligere slik at de vil inneholde andre ikke-proteinholdige deler som er kjent i faget og lett tilgjengelige. Fortrinnsvis er delene som er egnet for derivatisering av antistoffet vannløselige polymerer. Ikke-begrensende eksempler på vannløselige polymerer inkluderer, men er ikke begrenset til, polyetylenglykol (PEG), kopolymerer av etylenglykol/propylenglykol, karboksymetylcellulose, dekstran, polyvinylalkohol, polyvinylpyrrolidon, poly-1,3-dioksolan, poly-1,3,6-trioksan, etylen/maleinsyreanhydridkopolymer, polyaminosyrer (enten homopolymerer eller vilkårlige kopolymerer) og dekstran eller poly(n-vinylpyrrolidon)polyetylenglykol, polypropylenglykolhomopolymerer, polypropylenoksid/etylenoksid-kopolymerer, polyoksyetylerte polyoler (for eksempel glyserol), polyvinylalkohol og blandinger av disse. Polyetylenglykolpropionaldehyd kan ha fordeler under fremstilling fordi den er stabil i vann. Polymeren kan ha en hvilken som helst molekylvekt og kan være forgrenet eller uforgrenet. Antallet polymerer som er festet til antistoffet kan variere, og hvis mer enn én polymer er festet, kan de være de samme eller forskjellige molekyler. Generelt kan antallet og/eller typen av polymerer som anvendes for derivatisering bestemmes ut fra betraktninger som inkluderer, men ikke er begrenset til, de spesielle egenskapene eller funksjonene til antistoffet som skal forbedres, hvorvidt antistoffderivatet skal anvendes i en behandling under definerte forhold osv. Slike metoder og andre egnede formuleringer er offentliggjort i Remington: The Science and Practice of Pharmacy, 20. utg., Alfonso Gennaro, red., Philadelphia College of Pharmacy and Science (2000). C. Farmasøytiske formuleringer 30 3 Terapeutiske formuleringer fremstilles for lagring ved å blande den aktive bestanddelen med ønsket renhetsgrad med valgfrie farmasøytisk aksepterbare bærere, hjelpestoffer eller stabilisatorer (Remington: The Science and Practice of Pharmacy, 20. utg., Lippincott Williams & Wilkins, pub., Gennaro red., Philadelphia, PA 2000). Aksepterbare bærere, hjelpestoffer eller stabilisatorer er ikke-toksiske for mottakere ved doseringene og konsentrasjoner som benyttes, og inkluderer buffere, antioksidanter, inkludert askorbinsyre, metionin, vitamin E, natriummetabisulfitt; konserveringsmidler, isotonisitetsmidler, stabilisatorer, metallkomplekser (f.eks. Zn-proteinkomplekser); kelatdannere som f.eks. EDTA og/eller ikke-ioniske surfaktanter.
104 2 Hvis det terapeutiske midlet er et antistoffragment, foretrekkes det minste hemmende fragmentet som spesifikt binder bindingsdomenet av målproteinet. For eksempel kan det ut fra sekvensene til de variable regionene av antistoffet utformes antistoffragmenter eller til og med peptidmolekyler som beholder evnen til å binde målproteinsekvensen. Slike peptider kan syntetiseres kjemisk og/eller fremstilles ved å anvende rekombinant DNA-teknologi (se f.eks. Marasco et al., Proc. Natl. Acad. Sci. USA 90: [1993]). 1 Buffere anvendes til å kontrollere ph i et område som gir optimal terapeutisk virkning, særlig hvis stabiliteten er ph-avhengig. Buffere er fortrinnsvis til stede i konsentrasjoner som varierer fra rundt 0 mm til rundt 20 mm. Egnede buffermidler til anvendelse med den foreliggende oppfinnelsen inkluderer både organiske og uorganiske syrer og salter av disse. For eksempel sitrat, fosfat, suksinat, tartrat, fumarat, glukonat, oksalat, laktat, acetat. I tillegg kan buffere omfatte histidin og trimetylaminsalter, som f.eks. Tris Konserveringsmidler tilsettes for å hemme mikrobevekst, og er vanligvis til stede i et område fra 0,2 1,0 g/ml. Egnede konserveringsmidler til anvendelse med den foreliggende oppfinnelsen inkluderer oktadekyldimetylbenzylammoniumklorid; heksametoniumklorid; benzalkoniumhalogenider (for eksempel klorid, bromid, jodid), benzetoniumklorid; timerosal, fenol, butyleller benzylalkohol; alkylparabener som f.eks. metyl- eller propylparaben; katekol; resorcinol; sykloheksanol, 3-pentanol og m-kresol. Tonisitetsmidler, noen ganger omtalt som «stabilisatorer» er til stede for å justere eller opprettholde tonisiteten til væsken i en sammensetning. Når de anvendes til store, ladde biomolekyler, som f.eks. proteiner og antistoffer, kalles de ofte «stabilisatorer» fordi de kan interagere med de ladde gruppene i aminosyresidekjedene og derved minske faren for inter- og intramolekylære interaksjoner. Tonisitetsmidler kan være til stede i en hvilken som helst mengde mellom 0,1 vekt-% og 2 vekt-%, fortrinnsvis 1 til %, idet det tas hensyn til de relative mengdene av de andre bestanddelene. Foretrukne tonisitetsmidler inkluderer flerverdige sukkeralkoholer, fortrinnsvis trihydriske eller høyere sukkeralkoholer, som f.eks. glyserol, erytritol, arabitol, xylitol, sorbitol og mannitol.
105 3 1 Andre hjelpestoffer inkluderer midler som kan tjene som én av de følgende: (1) fyllmidler, (2) løseliggjørere, (3) stabilisatorer og (4) og midler som forhindrer denaturering eller tilklebing til beholderveggen. Slike hjelpestoffer inkluderer: polyhydriske sukkeralkoholer (spesifisert ovenfor); aminosyrer, som f.eks. alanin, glysin, glutamin, asparagin, histidin, arginin, lysin, ornitin, leucin, 2- fenylalanin, glutaminsyre, treonin osv.; organiske sukkerarter eller sukkeralkoholer som f.eks. sukrose, laktose, laktitol, trehalose, stakyose, mannose, sorbose, xylose, ribose, ribitol, myoinositose, myoinositol, galaktose, galaktitol, glyserol, syklitoler (f.eks. inositol), polyetylenglykol; svovelholdige reduksjonsmidler, som f.eks. urea, glutation, tioktinsyre, natriumtioglykolat, tioglyserol, α-monotioglyserol og natriumtiosulfat; proteiner med lav molekylvekt som f.eks. humant serumalbumin, bovint serumalbumin, gelatin eller andre immunoglobuliner; hydrofile polymerer, som f.eks. polyvinylpyrrolidon; monosakkarider (f.eks. xylose, mannose, fruktose, glukose, disakkarider (f.eks. laktose, maltose, sukrose); trisakkarider, som f.eks. raffinose; og polysakkarider, som f.eks. dekstrin eller dekstran Ikke-ioniske surfaktanter eller detergenter (også kjent som «fuktemidler») er til stede for å bidra til å løseliggjøre det terapeutiske midlet, samt for å beskytte det terapeutiske proteinet mot sammenklumping ved agitering, noe som også gjør det mulig for formuleringen å bli utsatt for overflateskjærstress uten at det aktive terapeutiske proteinet eller antistoffet denatureres. Ikkeioniske surfaktanter foreligger i et område på rundt 0,0 mg/ml til rundt 1,0 mg/ml, fortrinnsvis rundt 0,07 mg/ml til rundt 0,2 mg/ml. Egnede ikke-ioniske surfaktanter inkluderer polysorbater (20, 40, 60, 6, 80 osv.), polyoksamerer (184, 188 osv.), PLURONIC -polyoler, TRITON, polyoksyetylensorbitanmonoetere (TWEEN -20, TWEEN -80 osv.), lauromakrogol 400, polyoksyl-40-stearat, polyoksyetylenhydrogenert ricinusolje, 0 and 60, glyserolmonostearat, sukrosefettsyreester, metylcelluose og karboksymetylcellulose. Anioniske surfaktanter som kan anvendes inkluderer natriumlaurylsulfat, dioktylnatriumsulfosuksinat og dioktylnatriumsulfonat. Kationiske surfaktanter inkluderer benzalkoniumklorid eller benzetoniumklorid. 3 For at formuleringene skal kunne anvendes for in vivo-administrering, må de være sterile. Formuleringen kan bli gjort steril ved filtrering gjennom sterile filtreringsmembraner. De terapeutiske sammensetningene ifølge dette
106 4 dokumentet plasseres generelt i en beholder som har en steril adgangsport, for eksempel en pose for intravenøs løsning eller et glass med en propp som kan perforeres med en hypodermisk injeksjonsnål. Administreringsveien er i overensstemmelse med kjente og aksepterte metoder, for eksempel ved enkel eller multippel bolus eller infusjon over en lang tidsperiode på en egnet måte, for eksempel injeksjon eller infusjon gjennom subkutane, intravenøse, intraperitoneale, intramuskulære, intraarterielle, intralesjonale eller intraartikulære veier, topisk administrering, innånding eller ved metoder for langvarig frigjøring eller forlenget frigjøring. 1 Formuleringen i dette dokumentet kan også inneholde mer enn én aktiv forbindelse som er nødvendig for den spesielle indikasjonen som behandles, fortrinnsvis med komplementære aktiviteter som ikke påvirker hverandre ugunstig. Alternativt, eller i tillegg, kan sammensetningen omfatte et cytotoksisk middel, cytokin eller veksthemmende middel. Det er hensiktsmessig at slike molekyler er til stede i kombinasjon i mengder som er effektive for det tiltenkte formålet De aktive bestanddelene kan også innesluttes i mikrokapsler som fremstilles for eksempel ved konserveringsmetoder eller ved grenseflatepolymerisering, for eksempel henholdsvis mikrokapsler av hydroksymetylcellulose eller gelatin og poly-(metylmetakylat)mikrokapsler, i kolloidale legemiddelformuleringssystemer (for eksempel liposomer, albuminmikrokuler, mikroemulsjoner, nanopartikler og nanokapsler) eller i makroemulsjoner. Slike metoder er offentliggjort i Remington's Pharmaceutical Sciences 18. utgave, ovenfor Stabiliteten til proteinene og antistoffene som beskrives i dette dokumentet kan økes ved å anvende ikke-toksiske «vannløselige flerverdige metallsalter». Eksempler inkluderer Ga 2+, Mg 2+, Zn 2+, Fe 2+, Fe 3+, Cu 2+, Sn 2+, Sn 4+, Al 2+ og Al 3+. Eksempler på anioner som kan danne vannløselige salter med de ovennevnte flerverdige metallkationene inkluderer slike som er dannet av uorganiske syrer og/eller organiske syrer. Slike vannløselige salter har en løselighet i vann (ved 20 C) på minst rundt 20 mg/ml, alternativt minst rundt 0 mg/ml, alternativt minst rundt 200 mg/ml. Egnede uorganiske syrer som kan anvendes for å danne de «vannløselige flerverdige metallsaltene» inkluderer saltsyre, eddiksyre, svovelsyre, salpetersyre,
107 1 tiocyansyre og fosforsyre. Egnede organiske syrer som kan anvendes inkluderer alifatiske karboksylsyrer og aromatiske syrer. Alifatiske syrer innenfor denne definisjonen kan defineres som mettede eller umettede C 2-9 -karboksylsyrer (f.eks. alifatiske mono-, di- og trikarboksylsyrer). For eksempel inkluderer eksempler på monokarboksylsyrer innen denne definisjonen de mettede C monokarboksylsyrene eddiksyre, propionsyre, smørsyre, valeriansyre, kapronsyre, heptansyre, kaprylsyre, pelargonsyre og kaprinsyre og de umettede C monokarboksylsyrene akrylsyre, propriolsyre, metakrylsyre, krotonsyre og isokrotonsyre. Eksempler på dikarboksylsyrer inkluderer de mettede C dikarboksylsyrene malonsyre, ravsyre, glutarsyre, adipinsyre og pimelinsyre, mens umettede C 2-9 -dikarboksylsyrer inkluderer maleinsyre, fumarsyre, citrakonsyre og mesakonsyre. Eksempler på trikarboksylsyrer inkluderer de mettede C 2-9 -trikarboksylsyrene trikarballylsyre og 1,2,3-butantrikarboksylsyre. I tillegg kan karboksylsyrene ifølge denne definisjonen også inneholde én eller to hydroksylgrupper slik at det dannes hydroksykarboksylsyrer. Eksempler på hydroksykarboksylsyrer inkluderer glykolsyre, melkesyre, glyserinsyre, tartronsyre, eplesyre, vinsyre og sitronsyre. Aromatiske syrer innenfor denne definisjonen inkluderer benzosyre og salisylsyre Vanlige benyttede vannløselige flerverdige metallsalter som kan anvendes til å bidra til å stabilisere de innkapslede polypeptidene ifølge denne oppfinnelsen inkluderer for eksempel: (1) metallsaltene av uorganiske syrer: halogenider (for eksempel sinkklorid, kalsiumklorid), sulfater, nitrater, fosfater og tiocyanater; (2) de alifatiske karboksylsyremetallsaltene (f.eks. kalsiumacetat, sinkacetat, kalsiumpropionat, sinkglykolat, kalsiumlaktat, sinklaktat og sinktartrat); og (3) de aromatiske karboksylsyremetallsaltene benzoater (for eksempel sinkbenzoat) og salisylater. D. Behandlingsmetoder: 30 3 Til forebygging eller behandling av sykdom vil den hensiktsmessige dosen til et aktivt middel avhenge av typen sykdom som skal behandles som definert ovenfor, alvorlighetsgraden og forløpet av sykdommen, hvorvidt midlet administreres for forebyggende eller terapeutiske formål, tidligere behandling, pasientens kliniske historie og respons på midlet, og skjønn fra den behandlende legen. Det er hensiktsmessig at legemidlet administreres til pasienten enten på én gang eller gjennom en serie av behandlinger.
108 6 1 En foretrukket fremgangsmåte for behandling er behandling av IgE-formidlede forstyrrelser. IgE-formidlede forstyrrelser inkluderer atopiske forstyrrelser, som karakteriseres ved en arvelig tilbøyelighet til å respondere immunologisk på mange vanlige naturlig forekommende inhalerte og svelgede antigener og kontinuerlig produksjon av IgE-antistoffer. Spesifikke atopiske forstyrrelser inkluderer allergisk astma, allergisk rhinitt, atopisk dermatitt og allergisk gastroenteropati. Atopiske pasienter har ofte flere allergier, noe som betyr at de har IgE-antistoffer mot, og symptomer fra mange miljømessige allergener, inkludert pollen, sopp (f.eks. muggsopp), dyre- og insektrester og visse matvarer. Imidlertid er ikke forstyrrelser forbundet med forhøyede IgE-konsentrasjoner begrenset til de som har en arvelig (atopisk) etiologi. Andre forstyrrelser som er forbundet med forhøyet IgE-konsentrasjon, som synes å være IgEformidlede og lar seg behandle med formuleringene ifølge denne foreliggende oppfinnelsen inkluderer: overfølsomhet (f.eks. anafylaktisk overfølsomhet, eksem, elveblest, allergisk bronkopulmoær aspergillose, parasittsykdommer, hyper-ige-syndrom, ataksi-telangiektasi, Wiskott-Aldrich-syndrom, alymphoplasia thymica, IgE-myelom og transplantat-mot-vert-reaksjon Allergisk rhinitt, også kjent som allergisk rhinokonjunktivitt eller høysnue, er den mest vanlige manifestasjonen av en atopisk reaksjon på inhalerte allergener, alvorligheten og varigheten er ofte samsvarende med intensiteten og lengden til eksponeringen for allergenet. Det er en kronisk sykdom, som kan vise seg for første gang i alle aldre, men debuten er vanligvis i barndommen eller ungdomsårene. Et typisk anfall består av rikelig vannaktig rhinoré, krampaktig nysing, tett nese og kløe i nesen og ganen. Postnasal slimdrenering forårsaker også sår hals, kremting og hoste. Det kan også være symptomer på allergisk blefarokonjunktivitt, med intens kløe i øyets bindehinne og øyelokk, utslett, rennende øyne og lysskyhet. Alvorlige anfall ledsages ofte av systemisk uvelhet, slapphet, tretthet, og noen ganger stølhet i musklene etter intense nyseperioder. 3 Astma, også kjent som reversibel obstruktiv luftveissykdom, karakteriseres ved hyperreaktivitet i det trakeobronkiale treet overfor åndedrettsirritanter og luftrørsammentrekkende kjemikalier, som gir anfall av tungpustethet, dyspné, tetthet i brystet og hoste som er reversible spontant eller med behandling. Det er en kronisk sykdom som involverer alle luftveiene, men varierer i alvorlighetsgrad fra sporadiske milde forbigående episoder til alvorlig, kronisk,
109 7 livstruende bronkial obstruksjon. Astma og atopi kan eksistere sammen, men bare rundt halvparten av astmatikerne er også atopiske, og en enda mindre prosentandel av atopiske pasienter har også astma. Atopi og astma er imidlertid ikke helt uavhengige, siden astma forekommer oftere hos atopiske enn blant ikke-atopiske personer, særlig i barndommen. Videre har astma historisk blitt inndelt i to undergrupper, ytre astma og indre astma. 1 Ytre astma, også kjent som allergisk, atopisk eller immunologisk astma, er beskrivende for pasienter som generelt utvikler astma tidlig i livet, vanligvis i barndommen. Det eksisterer ofte samtidig andre manifestasjoner for atopi, inkludert eksem eller allergisk rhinitt. Astmaanfall kan forekomme i pollensesongen, i nærvær av dyr, eller ved eksponering for husstøv, fjærputer eller andre allergener. Hudtester viser positive hevelses- og utslettreaksjoner på de utløsende allergenene. Det er interessant at den totale IgEkonsentrasjonen i serum ofte er forhøyet, men noen ganger er normale Indre astma, også kjent som ikke-allergisk eller idiopatisk astma, forekommer vanligvis først i voksen alder, etter en tilsynelatende luftveisinfeksjon. Symptomer inkluderer kronisk eller tilbakevendende bronkial obstruksjon som ikke er relatert til pollensesongen eller eksponering for andre allergener. Hudtester er negative for de vanlige atopiske allergenene, IgE-konsentrasjonen i serum er normal. Andre symptomer inkluderer blodspytting og eosinofili. Andre skjemaer for å klassifisere astma i undergrupper, som acetylsalisylsyresensitiv, anstrengelsesutløst, smittsom og psykologisk definerer bare ytre utløsende faktorer som påvirker visse pasienter mer enn andre. 30 Endelig er det viktig å merke seg at mens noen klassifiseringer historisk bare har forbundet allergisk astma med IgE-avhengighet, er det nå sterke statistisk signifikante data som viser en sammenheng mellom IgE og astma (både allergisk og ikke-allergisk). Kapitel 27, «The Atopic Diseases», A.I. Terr i Medical Immunology, 9. utg., Simon and Schuster, Stites et al, red. (1997). Derfor inkluderer uttrykket «IgE-formidlede forstyrrelser» i denne patentsøknaden både allergisk og ikke-allergisk astma. 3 Fysiske tegn på et astmaanfall inkluderer takypné, hørbar hvesing, og anvendelse av hjelpemuskulaturen i åndingen. Rask puls og forhøyet blodtrykk er også vanligvis til stede, i likhet med forhøyet konsentrasjon av eosinofiler i
110 perifert blod og nasale sekreter. Lungefunksjonene viser en nedgang i strømningshastighet og 1 sekunds forsert ekspiratorisk volum (FEV 1 ). Den totale lungekapasiteten og funksjonelle restkapasiteten er vanligvis normal eller litt forhøyet, men kan være senket ved ekstrem bronkospasme. Patologien til astmaen kan skilles i tidlig- og senfasereaksjoner. Den tidlige fasen karakteriseres av sammentrekking av den glatte muskulaturen, ødem og hypersekresjon, mens senfasereaksjonene karakteriseres ved celleinflammasjon. Astma kan utløses av en rekke ikke-spesifikke faktorer, inkludert infeksjoner (f.eks. virusluftveisinfeksjoner), fysiologiske faktorer (f.eks. trening, hyperventilering, dyp pust, psykologiske faktorer), atmosfæriske faktorer (f.eks. svoveldioksid, ammoniakk, kald luft, ozon, destillert vanndamp), inntak av stoffer (for eksempel propranolol, acetylsalisylsyre, ikke-steroidiske antiinflammatoriske legemidler), eksperimentelle innåndede stoffer (for eksempel hypertone løsninger, sitronsyre, histamin, metakolin, prostaglandin F 2α ) og stoffer som pustes inn i yrkessammenheng (f.eks. isocyanater). Forskjellige andre yrkesmessige eller miljømessige allergener som forårsaker allergisk astma kan inkludere animalske produkter, insektstøv, sjødyr, planteprodukter, frukt, frø, blader og pollen, organiske fargestoffer og blekk, mikrober, enzymer, terapeutiske midler, steriliseringsmidler og uorganiske og organiske kjemikalier Atopisk dermatitt, også kjent som eksem, eksem, nevrodermatitt, atopisk eksem eller Besniers prurigo, er vanlig kronisk hudforstyrrelse som er spesifikk for en undergruppe av pasienter med de familiære og immunologiske trekkene ved atopi. Hovedtrekket er en kløende hudinflammatorisk respons, som utløser et karakteristisk symmetrisk fordelt hudutbrudd med tilbøyelighet til å ramme visse steder hyppigere. Ofte overproduseres også IgE av B-lymfocytter. Mens atopisk dermatitt klassifiseres som en kutan form for atopi fordi den er forbundet med allergisk rhinitt og astma og høy IgE-konsentrasjon, korrelerer imidlertid ikke alltid alvorlighetsgraden til dermatitten med eksponeringen for allergener ved hudtesting, og desensibilisering er (i motsetning til ved andre allergiske sykdommer) ikke effektiv behandling. Mens høy serum-ige er bekreftende for diagnose av allergisk astma, utelukkes det ikke av normale konsentrasjoner. Sykdommen kan vise seg for første gang i alle aldre, og lesjonene begynner akutt med erytematøs ødematøs papel eller plakk med skjelldannelse. Kløe fører til betenthet og skorpedannelse, deretter til kronisk lichenisering. På cellenivået er den akutte lesjonen ødemøs og dermis infiltrert med mononukleære celler,
111 9 CD4-lymfocytter. Nøytrofiler, eosinofiler, plasmaceller og basofiler er sjeldne, men degranulerte mastceller er til stede. Kroniske lesjoner har epidermal hyperplasi, hyperkeratose og parakeratose og dermis infiltreres med mononukleære celler, Langerhans-celler og mastceller. Det kan også være fokale områder med fibrose, inkludert involvering av perinevriumet til små nerver. 1 Allergisk gastroenteropati, også kjent som eosinofil gastroenteropati, er en uvanlig atopisk manifestasjon der flere IgE-matoverfølsomheter er forbundet med en lokal slimhinnereaksjon i mage-tarmkanalen. Den er sjelden hos voksne, mer vanlig, men forbigående, hos spedbarn. Tilstanden oppstår når de inntatte matallergenene reagerer med lokale IgE-antistoffer i jejunalslimhinnen som frigjør mastcelleformidlere, noe som gir gastrointestinale symptomer kort tid etter måltidet. Fortsatt eksponering gir kronisk inflammasjon, som fører til gastrointestinalt proteintap og hypoproteinemisk ødem. Blodtap gjennom den betente tarmslimhinnen kan være betydelig nok til å forårsake jernmangelanemi. Den allergiske reaksjonen skjer lokalt i den øvre gastrointestinale slimhinnen etter allergeneksponeringen, men går tilbake når allergenet unngås Anafylaksi og elveblest er klart IgE-formidlet, men de mangler genetiske determinanter og rammer ikke atopiske individer hyppigere. Anafylaksi er en akutt, generalisert allergisk reaksjon med samtidig involvering av flere organsystemer, vanligvis hjertet, luftveiene, huden og magen. Reaksjonen formidles immunologisk og forekommer ved eksponering for et allergen som individet tidligere er sensibilisert for. Elveblest og angioødem er fysisk hevelse, erytem og kløe som følge av histaminstimulerte reseptorer i overfladiske blodkar i huden, og er det typiske hudtrekket ved systemisk anafylaksi. Systemisk anafylaksi er opptreden av en IgEformidlet reaksjon samtidig i flere organer som følge av legemiddel, insektgift eller mat. Den forårsakes plutselig av allergenutløst, mastcellelastet IgE, som fører til dyp og livstruende endring i virkemåten til forskjellige vitale organer. Vaskulær kollaps, akutt luftveisobstruksjon, kutan vasodilatasjon og ødem, og gastrointestinale og urogenitale muskelspasmer forekommer nesten samtidig, men ikke alltid i samme grad. 3 Patologien for anafylaksi inkluderer angioødem og hyperutvidede lunger, med slimtetting av luftveiene og fokal atelektase. På cellenivået fremstår lungene på samme måte som i et akutt astmaanfall, med hypersekresjon fra bronkiale submukosale kjertler, mukøst og submukøst ødem, peribronkial blodstuvning
112 1 og eosinofili i bronkieveggene. Lungeødem og blødninger kan opptre. Bronkiale muskelspasmer, hyperinflasjon og til og med alveolruptur kan opptre. Viktige trekk ved human anafylaksi inkluderer ødem, blodstuvning og eosinofili i lamina propria i strupehodet, luftrøret, strupelokket og hypofarynks. Eksponering for allergenet kan skje ved innånding, injeksjon, innånding eller kontakt med huden eller slimhinnene. Reaksjonen begynner i løpet av sekunder eller minutter etter eksponeringen for allergenet. Det kan opptre en innledende skrekk eller følelse av forestående undergang, raskt fulgt av symptomer i ett eller flere målorgansystemer: hjertet, luftveiene, hud eller mage-tarmkanal Allergenene som er ansvarlige for anafylaksi er forskjellige fra de som vanligvis forbindes med atopi. Matvarer, legemidler, insektgifter eller lateks er de vanligste kildene. Matallergener inkluderer slike som finnes i krepsdyr, bløtdyr (f.eks. hummer, reker, krabbe), fisk, belgfrukter (f.eks. peanøtter, erter, bønner, lakris), frø (f.eks. sesam, bomullsfrø, karve, sennep, linfrø, solsikke), nøtter, bær, eggehvite, bokhvete og melk. Legemiddelallergenene inkluderer slike som finnes i heterologe proteiner og polypeptider, polysakkarider og hapteniske stoffer. Insektallergener inkluderer Hymenoptera-insekter, inkludert honningbie, veps, geithams og brannmaur. 2 Mens epinefrin er den vanlige behandlingen for anafylaksi, foreskrives det vanligvis antihistamin eller andre histaminblokkere for mindre alvorlig elveblest eller angioødemisk reaksjon. E. Kombinasjonsbehandling 30 Fremgangsmåten ifølge oppfinnelsen kan kombineres med kjente metoder for behandling av IgE-formidlet forstyrrelse, enten som kombinerte eller ekstra behandlingstrinn eller som ekstra komponenter i en terapeutisk formulering. 3 For eksempel kan antihistaminer, særlig ikke-sederende antihistaminer administreres før eller samtidig med antistoffene mot IgE ifølge oppfinnelsen. Egnede antihistaminer inkluderer antihistaminer til alkylamin (f.eks. klorfeniramin), etanolamin (f.eks. difenhydramin) og fenotiazin (f.eks. prometazin). Mange antihistaminer motvirker de farmakologiske virkningene av histamin ved å blokkere reseptorsetene deres på effektorcellene, mens andre
113 vanlige antihistaminlegemidler fungerer ved å blokkere histaminfrigjøringen fra mastceller som har blitt sensibilisert og armert med allergenspesifikk IgE (f.eks. kromolynnatrium). Eksempler på antihistaminer inkluderer astemizol, azatadinmaleat, brofeniraminmaleat, karbinoksaminmaleat, cetirizinhydroklorid, klemastinfumarat, cyproheptadinhydroklorid, deksbromfeniraminmaleat, deksklorfeniraminmaleat, dimenhydrinat, difenhydraminhydroklorid, doksylaminsuksinat, feksofenadinhydroklorid, terfenadinhydroklorid, hydroksyzinhydroklorid, loratadin, meklizinhydroklorid, tripelannaminsitrat, tripelennaminhydroklorid, triprolidinhydroklorid. Bestemte symptomer på IgE-formidlede forstyrrelser (f.eks. tidlige fasereaksjoner) kan bedres med sympatomimetika eller legemidler som har bronkodilatatorvirkning. Epinefrin er et bredtvirkende alfa- og betaadrenergikum som ofte administreres subkutant i en dose på 0,2 0, ml av en 1:0 vandig oppløsning. En form av epinefrin med mer langvarig virkning (dvs. terbutalin) i 1:200 suspensjon anvendes også når en mer langvarig virkning er ønsket. Andre egnede betaadrenergika inkluderer albuterol, pirbuterol, metaproterenol, salmeterol, isoetarin og formeterol for nasal administrering (f.eks. håndholdt forstøver, periodisk pusteapparat med positivt trykk eller trykkinhalatorer med utmålt dose) eller oralt. Bronkodilatasjonen kan også oppnås ved å administrere xantiner, særlig når de administreres i kombinasjon med de ovennevnte sympatomimetiske legemidlene. Eksempel på xantiner inkluderer aminofyllin (iv mg) og teofyllin (oralt, 20 µg/ml serumkonsentrasjon). Andre symptomer fra forskjellige IgE-formidlede forstyrrelser (for eksempel senfasereaksjoner) kan dempes ved behandling med glukokortikoider eller andre legemidler som har anti-inflammatorisk virkning. Prednison (30 60 mg daglig) administreres systemisk for alvorlige anfall, mens beklometasondipropionat, triamcinolonacetonid og flunisolid administreres i forstøvet form som langvarig vedlikeholdsbehandling. Andre kortikosteroider som har antiinflammatorisk virkning inkluderer: betametason, budesonid, deksametason, fludrokortisonacetat, flunisolid, flutikasonpropionat, hydrokortison, metylprednisolon, prednisolon, prednison, triamcinolon. Ikke-steroidiske anti-inflammatoriske legemidler som også kan anvendes i kombinasjon med de terapeutiske fremgangsmåtene ifølge oppfinnelsen inkluderer
114 acetaminofen, acetylsalisylsyre, bromfenaknatrium, diklofenaknatrium, diflunisal, etodolak, fenoprofenkalsium, flurbiprofen, ibuprofen, indometacin, ketoprofen, meklofenamatnatrium, mefenaminsyre, nabumeton, naproksen, naproksennatrium, oksyfenbutazon, fenylbutazon, piroksikam, sulindak, tolmetinnatrium. I tillegg kan den maksimale terapeutiske fordelen også oppnås med administrering av slimhinneavsvellende midler (f.eks. fenylefrin, fenylpropanolamin, pseudoefadrin), hosteundertrykkende midler (f.eks. dekstrometorfan, kodein eller hydrokodon) eller smertestillende midler (f.eks. acetaminofen, acetylsalisylsyre). Allergendesensibilisering er en behandlingsform der allergener injiseres til pasienten for å redusere eller eliminere den allergiske responsen. Den er også kjent som allergen immunterapi, hyposensibilisering eller allergivaksinasjon. Den anvendes ofte i kombinasjon med andre allergibehandlinger, men ikke ofte som en primær behandling. Den har blitt benyttet med hell når det er mulig å unngå allergener. En typisk allergendesensibiliseringsbehandling innebærer subkutan injeksjon av sterilt allergen i økende doser én eller to ganger i uken inntil det oppnås en dose som gir et forbigående lite lokalt område av inflammasjon ved injeksjonsstedet. Dosen gis så etter en vedlikeholdsplan én gang hver uke. Allergisk desensibilisering anvendes som oftest i behandling av allergisk astma og allergisk rhinitt, selv om den har hatt suksess ved behandling av anafylaksi. Desensibiliseringen har også blitt anvendt effektivt ved å anvende adjuvanser, som f.eks. ikke-komplett Freunds adjuvans, som er en emulsjon av vandig antigen i mineralolje. Den fysiologiske virkningen danner et uløselig væskedepot som det gradvis frigjøres dråper av allergenet fra. En annen form for allergendesensibilisering er å polymerisere monomere allergener med glutaraldehyd for å lage et molekyl med relativt lav allergenisitet (dvs. som forårsaker allergisk respons), og samtidig beholder en effektiv grad av immunogenisitet. 30 F. Farmasøytiske doseringer: 3 Doser og ønsket legemiddelkonsentrasjon av farmasøytiske sammensetninger ifølge den foreliggende oppfinnelsen kan variere med den spesielle anvendelsen man ser for seg. Bestemmelse av den egnede dosen eller administreringsveien ligger godt innenfor ferdigheten til en vanlig fagperson. Dyreforsøk gir pålitelig veiledning for å bestemme effektive doser for behandling av mennesker. Uspesifikk skalering av effektive doser kan utføres ifølge prinsippene som er
115 113 fastsatt av Mordenti, J. and Chappell, W. «The Use of Interspecies Scaling in Toxicokinetics,» i Toxicokinetics and New Drug Development, Yacobi et al., utg. Pergamon Press, New York 1989, s Når det anvendes in vivo-administrering av polypeptidene eller antistoffene som beskrives i dette dokumentet, kan normale doseringsmengder variere fra rundt ng/kg til rundt 0 mg/kg pattedyrkroppsvekt eller mer per dag, fortrinnsvis fra rundt 1 mg/kg/dag til mg/kg/dag, avhengig av administreringsveien. Veiledning om spesielle doser og leveringsfremgangsmåter er gitt i litteraturen, se for eksempel U.S. pat. nr ; ; eller Det er innenfor omfanget til oppfinnelsen at forskjellige formuleringer vil være effektive for forskjellige behandlinger og forskjellige forstyrrelser, og at administrering for å behandle et bestemt organ eller vev kan nødvendiggjøre formulering på en måte som er forskjellig fra formuleringen til et annet organ eller vev. Dessuten kan doseringene administreres enten ved én eller flere separate administreringer eller ved kontinuerlig infusjon. For gjentatt administrering gjennom flere dager eller lengre, avhengig av tilstanden, opprettholdes behandlingen inntil det opptrer en ønsket undertrykking av sykdomssymptomene. Andre doseringsregimer kan imidlertid være nyttige. Progresjonen av denne behandlingen overvåkes lett ved konvensjonelle metoder og analyser. G. Administrering av formuleringen 2 30 Formuleringene ifølge den foreliggende oppfinnelsen, inkludert, men ikke begrenset til rekonstituerte formuleringer, administreres til et pattedyr som har behov for behandling med proteinet, fortrinnsvis et menneske, i samsvar med kjente fremgangsmåter, som f.eks. intravenøs administrering som en bolus eller ved kontinuerlig infusjon i løpet en tidsperiode, ved intramuskulær, intraperitoneal, intracerebrospinal, subkutan, intraartikulær, intrasynovial, intratekal, oral, topisk eller inhalatorisk vei. 3 I foretrukne utførelsesformer administreres formuleringene til pattedyret ved subkutan (dvs. under huden) administrering. For slike formål kan formuleringen injiseres ved hjelp av en sprøyte. Andre anordninger for administrering av formuleringen er imidlertid tilgjengelig som f.eks. injeksjonsanordninger (f.eks. INJECT-EASE - og GENJECT -anordningene); injektorpenner (som f.eks.
116 114 GENPEN ); autoinjektoranordninger, nålfrie anordninger (f.eks. MEDIJECTOR og BIOJECTOR ); og subkutane plasterformuleringssystemer. I en spesifikk utførelsesform dreier den foreliggende oppfinnelsen seg om sett for en enkeltdoseadministreringsenhet. Slike sett omfatter en beholder med en vandig formulering av terapeutisk protein eller antistoff, inkludert både enkle eller forhåndsfylte sprøyter med flere kammer. Eksempler på forhåndsfylte sprøyter er tilgjengelige fra Vetter GmbH, Ravensburg, Tyskland Den egnede dosen («terapeutisk effektiv mengde») av proteinet vil være avhengig f.eks. av tilstanden som behandles, alvorlighetsgraden og forløpet av tilstanden, hvorvidt proteinet administreres for forebyggende eller terapeutiske formål, tidligere behandling, pasientens kliniske historie og respons på proteinet, type protein som anvendes og skjønn fra den behandlende legen. Det er hensiktsmessig at proteinet administreres til pasienten enten på én gang eller gjennom en serie av behandlinger og kan administreres til pasienten på et hvilket som helst tidspunkt fra diagnosen og senere. Proteinet kan administreres som den eneste behandlingen eller i kombinasjon med andre legemidler eller behandlinger som er nyttige ved behandling av den aktuelle tilstanden. Hvis det valgte proteinet er et antistoff, er fra rundt 0,1 til 20 mg/kg en første kandidatdose for administrering til pasienten, for eksempel enten ved én eller flere separate administreringer. Andre doseringsregimer kan imidlertid være nyttige. Progresjonen av denne behandlingen overvåkes lett ved konvensjonelle metoder. Anvendelser for en anti-ige-formulering (f.eks. rhumabe-2, rhmabe-26, Hu-901) inkluderer behandling eller forebygging av f.eks. IgE-formidlede allergiske sykdommer, parasittiske infeksjoner, interstitiell cystitt og astma. Avhengig av sykdommen eller forstyrrelsen som skal behandles administreres det en terapeutisk effektiv mengde (f.eks. fra rundt 1 til 1 mg/kg) av antistoffet mot IgE til pasienten. H. Produksjonsgjenstander 3 I en annen utførelsesform ifølge oppfinnelsen tilveiebringes en produksjonsgjenstand som inneholder formuleringen og fortrinnsvis tilveiebringer instruksjoner for anvendelsen. Produksjonsgjenstanden omfatter en beholder. Egnede beholdere inkluderer for eksempel flasker, glass (for
117 11 1 eksempel tokammerglass), sprøyter (som f.eks. en- eller tokammersprøyter) og reagensglass. Beholderen kan lages av en rekke forskjellige materialer, som f.eks. glass eller plast. Beholderen inneholder formuleringen. Etiketten, som er på eller forbundet med beholderen kan angi instruksjoner for rekonstituering og/eller anvendelse. Etiketten kan også indikere at formuleringen kan anvendes til eller er ment for subkutan administrering. Beholderen som inneholder formuleringen kan være et flerbruksglass som gjør det mulig å administrere den rekonstituerte formuleringen flere ganger (for eksempel 2 til 6 administreringer). Produksjonsgjenstanden kan også omfatte en andre beholder som omfatter et egnet fortynningsmiddel (f.eks. BWFI). Ved blanding av fortynningsmidlet og den frysetørkede formuleringen vil den endelige proteinkonsentrasjonen i den rekonstituerte formuleringen generelt være minst 0 mg/ml. Produksjonsgjenstanden kan dessuten inkludere andre materialer som er ønskelige fra et kommersielt og brukersynspunkt, inkludert andre buffere, fortynningsmidler, filtre, nåler, sprøyter og pakningsvedlegg med instruksjoner for bruk. 20 De følgende eksemplene kan gi en mer fullstendig forståelse av oppfinnelsen. De skal imidlertid ikke tolkes som begrensende for omfanget av oppfinnelsen. Alle sitatene i offentliggjøringen innføres herved uttrykkelig ved referanse. 2 I en annen utførelsesform tilveiebringer oppfinnelsen en produksjonsgjenstand som omfatter formuleringene som er beskrevet i dette dokumentet for administrering i en autoinjiseringsanordning. En autoinjektor kan beskrives som en injiseringsanordning som vil levere innholdet sitt ved aktivering uten at pasienten eller administratoren trenger å gjøre noe mer. De er spesielt egnet for selvmedisinering av terapeutiske formuleringer når leveringshastigheten må være konstant og leveringstiden er mer enn et lite øyeblikk. 30 Eksempel 1 3 Isolering av primat-ige Humant IgE ble klonet fra den humane IgE-myelomcellestammen U266 (ATCC, Manassas, VA, TIB #196). IgE-sekvensene fra rhesusape og krabbemakak ble bestemt ved å anvende kloning og sekvensering av IgE-et fra cdna i mononukleære celler i perifert blod fra rhesusaper og krabbemakaker, stimulert til de produserte IgE-vekslede cellene (PBMC). Forover- og
118 bakoverprimere (vist nedenfor) ble utformet fra de humane IgE-sekvensene. En oppstrøms foroverprimer ble utformet rett oppstrøms for CH2-domenet. Bakoverprimeren ble utformet i enden av transmembrandomenet. PBMC fra rhesusaper og krabbemakaker stammet fra California National Primate Research Center (Davis, CA). x 6 PBMC ble dyrket med IL-4 (0 ng/ml) og CD40L (3 µg/ml) (R&D Systems, Minneapolis, MN, henholdsvis #204-IL og #617-CL) i 4 dager. Cellene ble deretter høstet, RNA ble fremstilt (RNeasy Mini Kit, #746, Qiagen, Valencia, CA), og cdna fremstilt ved å anvende BD Sprint Powerscript oligo dt-primingssettet (BD Biosciences, San Jose, CA, #6398). Det ble så utført RT-PCR [94 C i 2 minutter; 94 C i 1 sekunder; 60 C i 30 sekunder; 68 C i 2 minutter; 30 sykluser; 68 C i minutter]. Taqpolymerase ble tilsatt ( minutter 72 C) etter de indikerte syklusene for å legge til A-overheng for TA-kloning (Invitrogen, Carlsbad, CA, #4-0641). PCRproduktene ble klonet ved TOPO-TA-kloning og klonene ble deretter sekvensverifisert (Genentech, Inc.). De humane sekvensene og rhesusape- og krabbemakaksekvensene ble sammenstilt ved hjelp av interne analyseprogrammer (Genentech, Inc.). De følgende homologiene er for sekvenser mellom CH2-domenet og de transmembrandomenet. Mellom rhesus (432 aminosyrer) og krabbemakak (431 aminosyrer) var det 6 forskjeller, som utgjør rundt 98,6 % homologi. Mellom menneske og rhesus var det 3 forskjeller som er 87,7 % homologi. Mellom menneske og krabbemakak var det 6 forskjeller, som er 87 % homologi (se figur 1). 2 Primere som anvendes til kloning: Foroverprimere oppstrøms for CH2-sekvensen: '-ccgcccaccgtgaagatcttacagtc (SEQ ID NO:63) Bakoverprimeren i enden av transmembrandomenet: '-cggctcgagactaggcgtggggctggaggacgttg (SEQ ID NO:64) 30 Eksempel 2 Isolering av anti-ige/m1' Abs 3 Generering av CH3-CH4-M1'/Fc-fusjonsproteinet Det humane IgE CH3-CH4-M1'/Fc-fusjonsproteinet ble dannet ved PCR-forsterking av de humane IgE CH3-CH4-M1'-domenene, i tillegg til ytterligere 1 aminosyrer umiddelbart nedstrøms for M1'-domenet fra cdna fremstilt av U266-celler (ATTC
119 117 Manassas, VA, TIB#196). RNA ble fremstilt av U266-celler (RNeasy Mini Kit, #746, Qiagen, Valencia, CA) og cdna fremstilt ved å anvende BD Sprint Powerscript oligo dt-priming (BD Biosciences, San Jose, CA, #6398). Det ble utført RT-PCR, og PCR-produktet ble klonet ved TOPO-TA-kloning (Invitrogen, Carlsbad, CA, #4-0641). Det ble lagt til en signalsekvens i N-enden ved PCRmutagenese for uttrykking i pattedyrceller og signalsekvens+ch3+ch4+m1 +1- aminosyrer ble klonet inn i en uttrykkingsvektor (prk huigg1 Fc; Genentech) for å lage en C-endefusjon med den humane IgG1 Fc-regionen. Sekvensen av det endelige proteinet, inkludert signalsekvensen og humant IgG1 Fc er som følger: 1 Proteinet ble fremstilt ved forbigående transfektering av CHO-celler (Genentech), og proteinet ble renset for cellekultursupernatant ved å anvende standard kolonnekromatografimetoder med protein A-sefarose Immunisering og danning av hybridomer Det ble laget et panel av panspesifikke antistoffer som selektivt binder M1'-delen av humant IgE-M1' ved hjelp av CH3-CH4-M1'/Fc-proteinet. Begge de bakre tråputene på en BALB/c-mus ble injisert med 1 µg CH3-CH4-M1'/Fc resuspendert i monofosforyl-lipid A og trehalosedikorynomykolat (MPL + TDM) adjuvans (Corixa, Hamilton, MT) ved 3- til 4-dagers intervall. Serum ble tatt etter 8 boosterinjeksjoner og titrert med enzymbundet immunsorbantanalyse (ELISA) og fluorescensaktivert cellesortering og flowcytometri (FACS-screening, se neste avsnitt) for å sikre at musen hadde god immunrespons mot CH3-CH4-M1'/Fc. Tre dager etter den endelige økningen ble knuteceller fra knehasen fusjonert med musemyelomcellestammen P3X63Ag.U.1 (se f.eks. Chuntharapai et al., 1997, Methods Enzymol. 288:1 27). Fusjonerte hybridomceller ble valgt fra ufusjonerte knehaseknuteceller eller myelomceller ved å anvende hypoksantinaminopterin-tymidin-seleksjon (HAT) i medium D fra ClonaCell
120 118 hybridomseleksjonssett (StemCell Technologies, Inc., Vancouver, BC, Canada), som fører til at det dannes 4224 kloner. 1 Screening for panspesifikke antihumane M1'-antistoffer Hybridomkloner laget som beskrevet tidligere ble screenet for produksjon av monoklonale antistoffer som binder til M1 -regionen i CH3-CH4-M1'/Fcproteinet, M1 -regionen til humant IgE-M1' som uttrykkes stabilt på overflaten av den murine B-cellelymfomcellestammen A20 (IgE-M1'/A20), og M1 - regionen til humant IgE-M1' som uttrykkes stabilt på overflaten av den BJABhumane B-cellelymfomcellestammen (IgE-M1'/BJAB). De negative kontrollene inkluderte humant CD-4/Fc, humant IgE som uttrykkes stabilt på overflaten av den murine B-cellelymfomcellestammen A20 (IgE/A20), den murine B- cellelymfomcellestammen A20 (A20), humant IgE som uttrykkes stabilt på overflaten av den BJAB-humane B-cellelymfomcellestammen (IgE/BJAB) og den BJAB-humane B-cellelymfomcellestammen (BJAB) ELISA-screening For å screene de 4224 klonene ble det utført enzymbundet immunosorbantanalyse (ELISA) generelt som beskrevet i Baker et al., 2002, Trends Biotechnol., 20: Analysene ble utført både på 384- og 96-brønners plater. Kort fortalt ble en 384-brønners (eller en 96-brønners) plate belagt med 0 µl kaprint antihumant IgG Fc (MP Biomedicals, Irvine, CA) i en konsentrasjon på 2 µg/ml i beleggingsbuffer (0,0 M karbonatbuffer, ph 9,6), forseglet og lagret over natten ved 4 C. Etter fjerning av beleggingsløsningen ble hver brønn tilsatt 80 µl (eller 200 µl i en 96-brønners plate) analyse-/blokkeringsløsning som inneholdt 0, % bovint serumalbumin og 0,0 % Tween -20 i PBS (ph 7,4) (ELISAfortynningsmiddel), og platene ble inkubert ved romtemperatur i én time med omrøring. Brønnene ble deretter vasket tre ganger med 0 µl (eller 300 µl for en 96-brønners plate) 0,0 % Tween -20 i PBS (vaskebuffer). Etter vasketrinnet ble det tilsatt 0 µl (eller 0 µl for en 96-brønners plate) av antigenløsningen som inneholdt 0,4 µg/ml CH3-CH4-M1'/Fc eller humant CD- 4/Fc i ELISA-fortynningsmiddel til hver brønn, og platene ble inkubert ved romtemperatur i én time med omrøring. Brønnene ble vasket tre ganger med vaskebuffer som før. Det ble tilsatt supernatant fra individuelle hybridomkloner slik at én brønn med CH3-CH4-M1'/Fc og én brønn med humant CD-4/Fc hver mottok 0 µl (eller 0 µl for en 96-brønners plate) med supernatant fra en
121 119 enkelt hybridomklon. Platene ble inkubert ved romtemperatur i én time med omrøring og brønnene ble vasket tre ganger med vaskebuffer som før. 1 Etter vaskingen ble 0 µl (eller 0 µl for en 96-brønners plate) av en 1:00 fortynning av ovint antimurint IgG koblet til pepperrotperoksidase (ingen kryssreaktivitet mot humant IgG (MP Biomedicals)) i ELISA-fortynningsmiddel tilsatt til hver brønn. Platene ble inkubert ved romtemperatur i én time med omrøring, vasket tre ganger med vaskebuffer som før og klappet tørre. Brønnene ble utviklet ved å tilsette 0 µl (eller 0 µl for en 96-brønners plate) tetrametylbenzidin (TMB) mikrobrønnperoksidasesubstrat (BioFX Laboratories, Owing Mills, MD, katalog #TMBW-00-01) til hver brønn og inkubere ved romtemperatur i minutter eller inntil det ble observert en god fargeforandring. Utviklingen ble stoppet ved å tilsette 0 µl (eller 0 µl for en 96-brønnrts plate) TMB stoppløsning (BioFX Laboratories katalog #BSTP-00-01) til hver brønn. Platene ble analysert med en Sunrise-plateleser (Tecan US, Inc., Research Triangle Park, NC) ved 60 nm Uimmunisert serum og polyserum ble anvendt som kontroller. De uimmuniserte prøvene inneholdt museserum før immunisering, og polyserum inneholdt murint antiserum etter immuniseringer. FACS-screening For å screene 3221-kloner som var lagd i eksempel 1 ble det utført fluorescensaktivert cellesortering (FACS) ved hjelp av IgE-M1'/A20- og IgE- M1'/BJAB-cellestammer. IgE/A20-, IgE/BJAB- A20 og BJAB-cellestammene fungerte som negative kontroller. Cellene ble resuspendert og sentrifugert ved 00 g i minutter ved 4 C. Mediet ble aspirert og cellene ble resuspendert i 4 C FACSFlow-buffer (BD Biosciences, San Jose, CA) med 1 % kalvefosterserum (cellefargingsbuffer). Cellene ble sentrifugert som før, mediet ble aspirert og cellene ble resuspendert med 2 x 6 celler/ml cellefargingsbuffer ved 4 C. Cellene ble lagt i 96-brønners rundbunnede plater med 0 µl/brønn (1x celler/brønn), og alle brønnene ble tilsatt 0 µl supernatant fra individuelle hybridomkloner slik at alle hybridomsupernatantene ble inkubert med en brønn som inneholdt hver av cellestammene. Platen ble inkubert på is i 30 minutter. Platen ble sentrifugert ved 00 g i minutter ved 4 C og supernatantene ble aspirert. Alle brønnene ble resuspendert i 200 µl cellefargingsbuffer ved 4 C, og platene ble sentrifugert som tidligere. Cellefargingsbufferen ble aspirert.
122 120 Etter vasketrinnet ble cellene i hver brønn suspendert på nytt i 0 µl av en 1:00-fortynning av kaprint antimurint IgG Fc bundet til R-fykoerytrin (Jackson Immunoresearch, West Grove, PA) i cellefargingsbuffer ved 4 C og platene ble inkubert i mørket på is i 30 minutter. Platen ble sentrifugert ved 00 g i minutter ved 4 C og supernatantene ble aspirert. Alle brønnene ble resuspendert i 200 µl cellefargingsbuffer ved 4 C, og platene ble sentrifugert som tidligere. Cellefargingsbufferen ble aspirert. Cellene i hver brønn ble resuspendert i 200 µl cellefargingsbuffer ved 4 C, og overført til mikrotiterrør på 1,2 ml (Quality Scientific Plastics, Petaluma, CA). FACS ble utført på en FACScan eller FACSCalibur (BD Biosciences) N-endesekvensering/isolering av RNA/sekvensering og kloning Antistoffene ble renset fra supernatanten av anti-ige/m1'-hybridomene, og ble analysert for N-endesekvensering for klonalitetsbestemmelse i fosfatbufret saltløsning. Alle antistoffene ble separert på en Precast 4-20 % Tris HCl SDS- PAGE (Invitrogen, Carlsbad, CA) under reduserende forhold. Den løste tunge kjeden (HC) og den lette kjeden (LC) ble begge utsatt for N-endesekvensering ved hjelp av en Procise 494 N-endesekvenserer (Applied Biosystems, Foster City, CA) ved å anvende 20-minutterssyklusen som beskrives av Henzel et al. Analytical Biochemistry 267, (1999). De første 2 restene ble sekvensert for å etablere monoklonalitet. Når HC- eller LC-kjeden til antistoffet var blokkert med en pyroglutamylgruppe som gjorde N-endeaminosyren utilgjengelig for Edman-sekvensering, ble den blokkerende gruppen fjernet før sekvenseringen. Pyroglutamylgruppen ble fjernet med pyroglutamataminopeptidase-enzymet (PGAP, Sigma, St Louis, MO) ved å anvende protokollen som er beskrevet av Pham et al. Electrophoresis 200, Sekvensen til de M1 -antistoffene med tunge (HC) og lette (LC) kjeder 7A6, 1C11, 47H4, 26A11, 4C1 og 28E9 ble bestemt til følgende: 7A6 HC QVQLQQSGAELVRPGASVTLSCKAS (SEQ ID NO:66) LC DIVMSQSPSSLTVSVGEKVTLSCKS (SEQ ID NO:67) 1C11
123 121 7A6 HC QVQLQQSGAELVRPGASVTLSCKAS (SEQ ID NO:68) LC DIVMSQSPSSLAVSVGEKVTMSCKS (SEQ ID NO:69) 47H4 HC EVKLVESGGGLVQPGGSRKLSCAAS (SEQ ID NO:70) LC DVVLTQTPLSLPVSLGDQASI (SEQ ID NO:71) 26A11 HC EVQLQQSGPELVKPGASVKMSCKAS (SEQ ID NO:72) LC DIQMTQTTSSLSASLGDRVTITCRS (SEQ ID NO:73) 4C1 HC QIQLVQSGPELKKPGETVK (SEQ ID NO:74) LC DVVMTQTPLTLSVTIGQPASISCK (SEQ ID NO:7) 28E9 HC EVKLVESGGGLVQPGGSLRLSCATS (SEQ ID NO:76) LC DIQMTQSPASLSVSVGETVTFTCR (SEQ ID NO:77) For å bestemme fullengdesekvensene ble '-degenererte primere utformet fra N-endeaminosyresekvensene og kjente Ig-gensegmenter, og det ble utført PCR med degenererte bevarte sekvenser for tunge og lette kjeder nedstrøms. Foroverprimere 7A6
124 122 7A6 HC FOR.BsiWI '-tcgacgtacgctcaggttcagctgcagcaatctggggctgagctgg (SEQ ID NO:78) LC FOR.EcoRV '-gatcgatatcgtgatgtcccagtctccctcctccctaac (SEQ ID NO:79) 1C11 HC FOR.BsiWI '-tcgacgtacgctcaggttcaattgcagcagtctggggctgagctgg (SEQ ID NO:80) LC FOR.EcoRV '-gatcgatatcgtaatgtctcagtctccttcctccctagc (SEQ ID NO:81) 47H4 HC 9.1HCF.BsiWI '-tcgacgtacgctgaggtgaagttggtggagtctgggggaggcttag (SEQ ID NO:82) LC 47H4LCF.EcoRV '-gatcgatatcgtgctgactcagactccactctccctgcc (SEQ ID NO:83) 26A11 HC G7F7HCF.BsiWI '-tcgacgtacgctgaggtccagctccagcagtctggacctgagc (SEQ ID NO:84) LC 2B4LCF.EcoRV '-gatcgatatccagatgacccaaactacatcctccctg (SEQ ID NO:8) 4C1 HC 2G6HCF.BsiWI '-tcgacgtacgctcagatccagttggtgcagtctggacctgagctg (SEQ ID NO:86) LC 9CLCF.EcoRV '-gatcgatatcgtgatgacgcagactccactcactttgtcgg (SEQ ID NO:87) 28E9
125 123 4C1 HC 9.1HCFBsiWI '-tcgacgtacgctgaggtgaagctggtggagtctgaaggaggcttgg (SEQ ID NO:88) LC E.LCF.EcoRV '-gatcgatatccagatgcccagtctccagcctccctatc (SEQ ID NO:89) Bakoverprimere Tungkjedeprimer (HCR) '-ctggacagggatccagagttccaggtcactgtcactggctcaggg ID NO:90) (SEQ Lettkjedeprimer (LCR) '-ctgtaggtgctgtctttgctgtcctgatcagtccaactg (SEQ ID NO:91) 1 20 RNA ble høstet fra anti-ige/m1'-hybridomene (RNeasy Mini Kit, #746, Qiagen, Valencia, CA), og cdna fremstilt ved å anvende BD Sprint Powerscript oligo-dtprimingssett (BD Biosciences, San Jose, CA, #6398). PCR ble utført ved å anvende de ovennevnte primerne, og PCR-produktene ble klonet ved å anvende et TOPO-TA-sett (Invitrogen, Carlsbad, CA, #4-0641). PCR ble utført ved hjelp av Platinum Pfx High Fidelity Polymerase (Invitrogen, Carlsbad, CA, # ) med PCR-forhold [94 C i 2 minutter; 94 C i 1 sekunder; 60 C i 30 sekunder; 68 C i 2 minutter; 30 sykluser; 68 C i minutter]. Klonene ble screenet for innsettinger og flere kloner ble sekvensert (Genentech, Inc.) for å bekrefte den opprinnelige gensekvensen for den tunge og lette kjeden i anti-ige/m1. Variable domener fra antistoff mot M1 ble subklonet inn i uttrykkingsvektorer med forskjellige Fc-regioner for å lage kimære antistoffer med forskjellige arter og/eller isotype-fc. Primere ble utviklet for å klone det variable domenet (CDR + ramme) til murint IgG2a, murint IgG2a-DANA og humane Fc-regioner fra IgG1-Fc. Primerne ble utformet for å innføre restriksjonsseter med tung- og lettkjedesekvens. De tunge kjedene ble klonet ved hjelp av BsiWI og ApaI. De lette kjedene ble klonet ved hjelp av EcoRV og KpnI. De nye PCR-produktene ble så digerert og bundet i migg2a (LPG), migg2a-dana (perk-e27dana) eller
126 124 huigg1 for tung kjede, eller mkappa- (LPG2) eller hukappa-uttrykkingsvektorene (Genentech, Inc.). De endelige klonene ble sekvensverifisert (Genentech, Inc.). 3'-primere og forover- og bakoverprimerkombinasjonene for kloning av variable CDR-sekvenser er som følger: Bakoverprimere 7A6 HC REV.ApaI '-accgatgggcccttggtggaggctgaagagactgtgag (SEQ ID NO:92) LC REV.KpnI '-ccttggtaccccctccgaacgtgtacggatagctataatattg (SEQ ID NO:93) 1C11 HC REV.ApaI '-gaccgatgggcccttggtggaggctgaggagactgtg (SEQ ID NO:94) LC REV.KpnI '-ccttggtaccccctccgaacgtgtacggatagctataa (SEQ ID NO:9) 47H4 HC 47H4HCR.ApaI '-tgggcccttggtggaggctgaggagacggtgactgag (SEQ ID NO:96) LC 47H4LCR.KpnI '-ttccaacttggtacctccacc (SEQ ID NO:97) 26A11 HC 7G8HCR.ApaI '- gaccgatgggcccttggtggargctgcagagacagtgaccagag (SEQ ID NO:98) LC 47H4LCR.KpnI '-ttccaacttggtacctccacc (SEQ ID NO:99) 4C1
127 12 4C1 HC 4C1HCR.ApaI '-cgatgggcccttggtggargckgaggagacggtgagaatg (SEQ ID NO:0) LC C2LCR.KpnI '-agcttggtaccccctccg (SEQ ID NO:1) 28E9 HC 28E9.HC.REV.ApaI '-cgatgggcccttggtggaggctgaggagacggcgactgag (SEQ ID NO:2) LC 1D.2LCR,KpnI '- ttccaacttggtacccgagccg (SEQ ID NO:3) Primerkombinasjoner: Forover Bakover 7A6: HC 7A6.FOR.BsiWI --- 7A6.REV.ApaI LC 7A6.FOR.EcoRV --- 7A6.REV.KpnI 1C11: HC 1C11.FOR.BsiWI --- 1C11.REV.ApaI LC 1C11.FOR.EcoRV --- 1C11.REV.KpnI 47H4: HC 9.1.HCF.BsiWI --- 7H4HCR.ApaI LC* 47H4.LCF.EcoRV H4.LCR.KpnI 26A11 HC C7F7.HCF.BsiWI --- 7G8.HCR.ApaI LC 2B4.LCF.EcoRV H4.LCR.KpnI 4C1 HC 2G6.HCF.BsiWI --- 4C1.HCR.ApaI LC 9C.LCF.EcoRV --- C2.LCR.KpnI 28E9 HC 9.1.HCF.BsiWI E9.HCR.ApaI
128 126 Forover Bakover LC E.LCF.EcoRV -- 1D2.LCR.KpnI Den lette kjeden til 47H4 hadde et indre KpnI-sete som utelukket direkte kloning inn i mkappa- og hukappa-uttrykkingssvektorene. Den indre KpnI-setet ble mutert ved hjelp av Quick Change XL Site Directed Mutagenesis Kit (Stratagene, Cedar Creek, Texas, #20017-) [9 C i 1 minutt; 9 C i 0 sekunder; 60 C i 0 sekunder; 68 C i 4, minutter; 68 C i 7 minutter]. Primere ble utviklet ifølge kravene i settet for å mutere et enkelt nukleotid i KpnI-setet som ikke endret aminosyresekvensen. TAC ble mutert til TAT som bevarte tyrosinet (Y) i denne posisjonen. De endelige klonene ble sekvensverifisert (Genentech, Inc.). Primerne som ble anvendt til mutagenesen var: FOR '-cc tat tta cat tgg tat ctg cag aag cca ggc c (SEQ ID NO:4) REV '-ggc ctg gct tct gca gat acc aat gta aat agg (SEQ ID NO:) 1 Eksempel 2A 20 2 Humanisering av antistoffer mot IgE/M1' Dette eksemplet beskriver humanisering av de murine antistoffene 26A11, 7A6 og 47H4 mot IgE/M1'. Materialer og fremgangsmåter M1'-domenet til membranforbundet humant IgE ble uttrykt som en Fc-fusjon i CHO-celler og renset ved konvensjonelle midler. Hybridomene som uttrykte antistoffene 26A11, 7A6 og 47H4 ble fremstilt ved immunisering av mus med det rekombinante M1'-Fc-fusjonsproteinet og identifisert med ELISA ved hjelp av plater belagt med M1'-Fc. Funksjonelle antistoffer ble identifisert ved evnen deres til å binde seg til de M1'-uttrykkende cellene og fremme apoptose. 30 Kloning av variable domener fra murint 26A11, 7A6 og 47H4 Total RNA ble tatt fra hybridomceller som produserte 26A11, 7A6 eller 47H4 ved standard fremgangsmåter. Det variable lette (VL) og variable tunge (VH) domenet ble forsterket ved å anvende RT-PCR med degenererte primere på den tunge og den lette kjeden. Foroverprimerne var spesifikke for N- endeaminosyresekvensen til VL- og VH-regionene. LC- og HC-reversprimerne
129 127 ble utformet for å anløpes til en region i det konstante lette domenet (CL) og det konstante tunge domenet 1 (CH1), som er svært godt bevart hos flere arter. Polynukleotidsekvensen til innsettingene ble bestemt ved rutinemessige sekvenseringsfremgangsmåter. VL- og VH-sekvensene til 26A11, 7A6 og 47H4 er vist i figur 6A og 6D, 6B og 6E og 6C og 6F respektive. Direkte poding av hypervariable regioner på akseptoren human konsensusramme Fagemidet som anvendes til dette arbeidet er en enverdig Fab-g3-displayvektor som består av 2 åpne leserammer under kontroll av en enkelt phoa-promotor. Den første åpne leserammen består av stiisignalsekvensen fusjonert til VL- og CH1-domenene til akseptorens lette kjede og den andre består av stii-signalsekvensen fusjonert til VH- og CH1-domenene til akseptorens tunge kjede, fulgt av det mindre bakteriofagkappeproteinet P VL- og VH-domenene fra de murine 26A11-, 7A6- eller 47H4-antistoffene (mu26a11, mu7a6 eller mu47h4) ble sammenstilt med de humane konsensussekvensene VL kappa I (huki) og human VH, undergruppe III (huiii). For å fremstille CDR-podingene ble hypervariable regioner fra mu26a11, mu7a6 eller mu47h4 podet inn i huki- og huiii-konsensusakseptorrammer for å lage den direkte CDR-podingen (26A11.v1, 7A6.v1 eller 47H4.v1) (figur 6A 6F). I VLdomenet ble de følgende regionene podet til den humane konsensusakseptoren: posisjon (L1), 0 6 (L2) og (L3). I VH-domenet ble posisjon 26 3 (H1), 49 6 (H2) og 94 2 (H3) podet. MacCallum et al. [MacCallum et al. J. Mol. Biol. 262: (1996))] har analysert krystallstrukturene til antistoff- og antigenkomplekser og funnet at posisjon 49 og 94 i den tunge kjeden er en del av kontaktregionen, dermed synes det rimelig å inkludere disse posisjonene i definisjonen av CDR-H2 og CDR-H3 ved humanisering av antistoffer De humaniserte antistoffene 26A11.v1, 7A6.v1 og 47H4.v1 mot IgE/M1' ble dannet som IgG-antistoffer ved Kunkel-mutagenese av LC- og HCuttrykkingsvektorene ved å anvende separate oligonukleotider for hver hypervariable region. Aminosyreendringer for å øke stabiliteten ble også gjort ved Kunkel-mutagenese. Kunkel et al., J. Biol. Chem. 263(29): (1988). De korrekte klonene ble identifisert ved DNA-sekvensering. IgG-produksjon For screening ble IgG-varianter innledningsvis fremstilt i 293-celler. Vektorer som koder for VL og VH (2 µg) ble transfektert i 293-celler
130 128 ved hjelp av FuGene-systemet. 00 µl av FuGene ble blandet med 4, ml DMEMmedium som ikke inneholdt FBS og inkubert ved romtemperatur i min. Hver kjede (2 µg) ble tilsatt til denne blandingen og inkubert ved romtemperatur i 20 min og deretter overført til fem T--kolber for transfektering over natten ved 37 C i % CO 2. Dagen etter ble mediet som inneholdt transfeksjonsblandingen fjernet og erstattet med 23 ml PS04-medium med 0,1 ml/l sporelementer (A0934) og mg/l insulin (A0940). Cellene ble inkubert i ytterligere dager og så ble mediet høstet ved 00 rpm i min og sterilt filtrert med et lite proteinbindende filter på 0,22 µm. Prøvene kunne lagres ved 4 C etter tilsetting av 2, ml 0,1 % PMSF for hver 12 ml av mediet Affinitetsbestemmelser Affinitetsbestemmelsene ble utført ved Scatchardanalyse, en cellebindende ELISA og ved overflateplasmonresonans ved hjelp av BIAcore eller A0. Scatchard-analyse - IgE/M1 -celler fra menneske (Daudi), rhesusape og krabbemakak ble fremstilt for binding i kald bindingsbuffer som besto av basismedium med mm HEPES ph 7,4, og 2 % FBS samt 40 µg/ml humant IgG. Cellene blir fortynnet til en konsentrasjon på 1,7 x 6 celler/ml og oppbevart på is inntil de tilsettes til analysen. Proteinene/antistoffene blir jodert med Iodogenmetoden. Forskjellige konsentrasjoner av kaldt protein fremstilles i triplikat ved hjelp av en 1:2-fortynning som starter med metningskonsentrasjonen og slutter ved en konsentrasjon på null, med totalt 14 konsentrasjoner. Alle fortynningene blir tilsatt varmt protein med en enkelt konsentrasjon for å konkurrere med det kalde proteinet. Til slutt blir cellene tilsatt til blandingen av de varme og kalde proteinene; det ble brukt celler per prøve. Analyseblandingen ble oppbevart og ristet ved 4 C i 4 timer. Alle prøvene blir så filtrert på membranen, vasket minst 3 ganger med bindingsbuffer, satt til tørking og så telt i 1 minutt med Perkin Elmer Wizard 1470 Auto Gamma Counter. Konkurransecellebindingsanalyse ved elektrokjemiluminescens De relative bindingsaffinitetene til humaniserte varianter ble målt ved en konkurransecellebindingsanalyse ved elektrokjemiluminescens: Transfekterte BJAB-celler som uttrykte IgE-M1' (BJAB-IgE/M1') (lange) eller kontrolltransfekterte celler som ikke uttrykte M1' (BJAB-IgE) (kort) ble vasket med fosfatbufret saltløsning (PBS), og utsådd med celler/brønn i 2 µl PBS på 96-brønners MULTI-ARRAY High Bind-plater (Meso Scale Discovery). Cellene ble inkubert på platene i én time ved
131 romtemperatur for å la cellene feste seg til brønnene. For å blokkere ikke-spesifikk binding ble det tilsatt 2 µl av 30 % FBS i PBS til hver brønn. Platene ble deretter inkubert ved romtemperatur i 30 minutter med forsiktig risting. Blokkeringsløsningen ble dekantert og tilsatt seriefortynninger av humaniserte varianter (0, nm) supplementert med en fast mengde av biotinylert antistoff mot IgE M1' (33,3 nm 26A11 eller 47H4) i 2 µl PBS som inneholdt 2 % FBS (analysebuffer). Etter én times inkubering ved romtemperatur med svak omrøring ble platene vasket tre ganger med PBS (300 µl/brønn) ved hjelp av en mikroplateskive (ELx40 Select, Bio-Tek Instruments, Inc., Winooski, VT). De bundne antistoffene ble detektert ved å tilsette 2 µl av 0,2 µg/ml ruteniummerket streptavidin i analysebuffer. Etter en én times inkubering ved romtemperatur ble platene vasket og tilsatt Tris-basert Read Buffer T (1x, uten surfaktant) (Meso Scale Discovery) ( µl/brønn). Elektrokjemiluminescenssignaler ble registrert med Sector Imager 6000-leseren (Meso Scale Discovery). Biacore 2000 To orienteringer ble anvendt; enten ble M1'-Fc eller en bestemt antistoffvariant immobilisert (rundt 0 RU, i mm natriumacetat ved ph 4,8 på en CM-sensorbrikke) og den tilsvarende liganden (henholdsvis antistoffvariant eller M1 Fc) var analytten (injisert med en hastighet på 30 µl/min, og en 2-gangers seriefortynning på 0, til 00 nm i PBST). Hver prøve ble analysert med 3-minutters assosiering og -minutters dissosiering. Etter hver injeksjon ble brikken regenerert med mm glycin ved ph 1,7. 2 Biacore A-0 M1' ble immobilisert (rundt 0 RU, i mm natriumacetat ved ph 4,8 på en CM-sensorbrikke) og antistoffvarianten var analytten (injisert med en hastighet på 30 µl/min, og en 2-gangers seriefortynning på 0, til 00 nm i PBST). Hver prøve ble analysert med -minutters assosiering og -minutters dissosiering. Etter hver injeksjon ble brikken regenerert med mm glycin ved ph 1,7. 30 Bindingsresponsen ble korrigert ved å trekke en kontrollflowcelle fra flowceller med 18F7-variant-IgG. En 1:1-Languirmodell av samtidig tilpasning av k on og k off ble anvendt til kinetikkanalyse. 3 Resultater og diskusjon CDR-poding av 26A11, 7A6 og 47H4 - Den humane akseptorrammen som ble anvendt for humaniseringen bygger på det humane kappa-i- VL-konsensusdomenet og det humane VH-konsensusdomenet, undergruppe III. VL- og VH-domenene til
132 mu26a11, mu7a6 og mu47h4 ble sammenstilt med de humane kappa I- og undergruppe-iii-domenene; hver komplementaritetsbestemmende region (CDR) ble identifisert og podet inn i den humane akseptorrammen for å lage et CDRtransplantat som kunne uttrykkes som et IgG (figur 6A 6F). Affinitetsevaluering - 26A11.v1, 7A6.v1 og 47H4.v1 ble evaluert ved cellebindings-elisa og ved overflateplasmonresonans (tabell 1). Disse analysene tydet på at CDR-transplantatene hadde affiniteter som var svært like de respektive hybridomantistoffene deres. Eliminering av potensielle deamideringsseter og isoasparaginsyredannende seter i CDR-L1 av 26A11 og 47H4 - For å unngå potensielle fremstillingsproblemer ble de potensielle isoasparaginsyredannende setene Asn 28 -Gly 29 i CDR-L1 av 47H4.v1 og Asn 31 -Ser 32 i CDR-L1 av 26A11.v1 eliminert ved å ta prøve av rester som ble funnet i andre antistoffer ved disse posisjonene (figur 6A, 6C, tabell I). Endring av Gly 29 til Ala 29 i CDR-L1 av 47H4.v1 (47H4.v2 og 47H4.v), og Ser 32 til Tyr 32 i CDR-L1 av 26A11 (26A11.v3 og 26A11.v6) eller Ala 32 (26A11.v2 eller 26A.11.v) viste seg å være egnede substitusjoner. I tillegg tjente også endring av Asn 31 til enten Ser 31 (26A11.v13 eller 26A11.v1) eller Gln 31 (26A11.v14 eller 26A11.v16) til å eliminere potensiell deamidering og isoasparaginsyredanning og ga kandidater med affiniteter tilsvarende de respektive hybridomantistoffene deres. 2 Potensiell oksidering av Met 34 i CDR-H1 av enten 26A11.v1 eller 47H4.v1 ble unngått ved å endre denne resten til Ile 34 i begge antistoffene uten å påvirke M1'-bindingen. Eksempel Spesifisiteten til antistoffer mot M1' Spesifisiteten til antistoffkloner mot IgE/M1' ble testet på den humane B- cellestammen BJAB (ATCC Manassas, VA, #HB-136) transfektert med IgE med M1'-sekvensen («lang») eller IgE uten M1'-sekvensen («kortform»). IgE BJABcellestammene ble produsert ved retroviral transduksjon av BJAB-celler ved hjelp av en pmscv uttrykkingsvektor (Washington University, St. Louis, MO). cdna-et til den tunge og lette fullengdekjeden til den humane IgE-B-cellereseptoren ble tatt fra den humane IgE-myelomcellestammen U266 (ATCC, Manassas, VA,
133 TIB#196), og ble subklonet inn i den retrovirale vektoren i kombinasjon med en IRES (Internal Ribosomal Entry Sequence) for å gi mulighet for bicistronisk kotransfektering og kouttrykking av den tunge og lette antistoffkjeden. Nærmere beskrivelse av fremgangsmåten for retroviral transduksjon er gitt nedenfor. PG13-pakkeceller (ATCC CRL-686) ble utsådd på en cm vevskulturplate med 2 x 6 celler per plate (DMEM høy glukose, % FBS, penicillin, streptomycin, 2 mm L-glutamin) i 24 timer. Cellene ble transfektert med pmscv- DNA-konstruksjoner ved hjelp av FuGENE 6 og dyrket i 2 dager ved 37 C med % CO 2. En cellekultursupernatant som inneholdt retroviruspartiklene ble høstet og filtrert gjennom et 0,4 mikron-filter. Sterilt protaminsulfat ble tilsatt til en sluttkonsentrasjon på µg/ml og 4 ml av supernatanten ble brukt til å infisere rundt 1 x 6 BJAB-celler ved rotasjonsinfisering ved 32 C i 90 minutter, fulgt av fortsatt dyrking i retrovirussupernatanten i 3 4 timer ved 37 C i % CO 2. De infiserte BJAB-cellene ble utvunnet, overført til RPMI-cellekulturmedium (RPMI, % FBS (Hyclone, Logun, UT, #SH ), penicillin, streptomycin, 2 mm L-glutamin), og ekspandert for sortering. De positivt transfekterte cellene ble identifisert ved flowcytometri ved å anvende MAE11 (Genentech) antistoff mot humant IgE til å detektere overflate-ige. Antistoffer mot IgE/M1' er spesifikke for den lange formen og ikke kortformen av membran-ige ifølge flowcytometrisk farging av BJAB-cellestammer med kort og lang form av IgE. Antistoffet mot humant IgE (Mae11) (Genentech, Inc.) ble anvendt som en positiv kontroll på grunn av evnen til å binde både den lange og korte formen av IgE. Antistoffer mot gp120 migg1 eller mot migg2a fra beiskambrosia ble anvendt som isotypekontroller (Genentech, Inc) BJAB-cellene ble dyrket i RPMI % FBS (Hyclone, Logun, UT, #SH ), penicillin/streptomycin, 2 mm L-glutamin (Genentech, Inc.). 0, x 6 BJAB langeller BJAB kort-celler ble blokkert med museserum ( µl) (VWR, #RLD8-00) og humant serum ( µl) (Sigma, St. Louis, MO, #S-227) i 1 minutter på is i 0 µl FACS-vaskebuffer (2 % FBS i 1X PBS (Genentech, Inc.). Deretter ble cellene farget med 1 µg/ml av hvert av antistoffene mot M1' i ytterligere 20 minutter på is og deretter vasket med 1 ml FACS-vaskebuffer og sentrifugert ved 1200 rpm i minutter. Cellene ble resuspendert i 0 µl FACS-vaskebuffer og deretter farget med kaprint antimurint IgG-PE ( µl) (Caltag/Invitrogen, Carlsbad, CA, #M ) på is i 20 minutter. Cellene ble vasket som tidligere og
134 132 1 resuspendert i 1 ml FACS-vaskebuffer for analyse på FACSCalibur-maskinen (BD, Inc., Franklin Lakes, NJ). Alle de testede antistoffene var spesifikke for den lange formen av IgE og bandt ikke BJAB-IgE/kort-celler mer enn isotypekontrollen. Vi har observert en lang rekke relative affiniteter ut fra fargeintensitetene: Vi har også vurdert binding av antistoff mot M1' til U266-cellestammen (ATCC, Manassas, VA, #TIB196) som naturlig uttrykker lav konsentrasjon av humant IgE-holdig M1' på celleoverflaten. U266-cellene oppbevares ved høy tetthet (1 2 x 6 /ml) i hybridommedium [RPMI 1640, 1 % kalvefosterserum, 2 mm L- glutamin, mm HEPES, 4, g/l glukose, 1, g/l bikarbonat]. U266-cellene ble farget med 1 µg/ml antistoffer mot M'. 47H4, 47G4 og 42A farget U266 ved liknende konsentrasjoner som Mae11. Antistoffene 42H4, 4C1 og 28E9 farget U266 ved svært lav konsentrasjon. 7A6, 1C11, 26A11, 1D2 og 26B11 farget ikke U266. (Figur 2C) Eksempel 3A 20 Bindingsspesifisiteten til murint antistoff mot IgE/M1' Figur 2D F viser bindingen for de murine antistoffene mot IgE/M1' 47H4, 26A11 og 7A6 til en samling cellestammer som uttrykker IgE/M1 fra menneske, rhesusape og krabbemakak Spesifisiteten til kloner av antistoff mot IgE/M1' ble testet på den humane B- cellestammen BJAB (ATCC Manassas, VA, #HB-136) og Daudi (ATCC #CCL-213) infisert med retrovirus for å uttrykke den lange formen av IgE med M1'-sekvensen eller den korte formen av IgE som mangler M1'-sekvensen. BJAB-cellene ble dyrket i RPMI % FBS (Hyclone, Logun, UT, #SH ), penicillin/streptomycin, 2 mm L-glutamin (Genentech, Inc.). 0, x 6 BJAB langeller BJAB kort-celler ble blokkert med museserum ( µl) (VWR, #RLD8-00) og humant serum ( µl) (Sigma, St. Louis, MO, #S-227) i 1 minutter på is i 0 µl FACS-vaskebuffer (2 % FBS i 1X PBS (Genentech, Inc.). Cellene ble deretter farget med 1 µg/ml av hvert av antistoffene mot M1' i ytterligere 20 minutter på is og deretter vasket med 1 ml FACS-vaskebuffer og sentrifugert ved 1200 rpm i minutter. Cellene ble resuspendert i 0 µl FACS-vaskebuffer og deretter farget med kaprint antimurint IgG-PE ( µl) (Caltag/Invitrogen, Carlsbad, CA, #M ) på is i 20 minutter. Cellene ble vasket som tidligere og resuspendert i 1 ml FACS-vaskebuffer for analyse på FACSCalibur-maskinen (BD,
135 133 Inc., Franklin Lakes, NJ). Alle de testede antistoffene var spesifikke for den lange formen av IgE og bandt ikke BJAB-IgE/kort-celler mer enn isotypekontrollen. Vi observerte en lang rekke relative affiniteter ut fra fargingsintensitetene. Spesifisiteten til kloner av antistoff mot IgE/M1' ble testet på den humane B- cellestammen Daudi infisert ved retrovirus for å uttrykke membran IgE med M1'-sekvensen (lang form) eller et membran-ige som mangler M1'-sekvensen (kort form). Antistoffene mot IgE/M1' (47H4, 26A11, 7A6) er spesifikke for den lange formen og ikke den korte formen (figur 2D). Et antistoff mot gp120 migg1 ble anvendt som isotypekontroll (Genentech, Inc). 1 Den humane B-cellestammen Daudi ble transdusert med retroviruset som inneholdt den humane IgE/primat-M1'-kassetten for IgE/M1 enten fra rhesus, krabbemakak eller menneske. Etter transduksjonen ble celler som uttrykker humant IgE/M1, rhesusape-ige/m1' eller krabbemakak-ige/m1' sortert med en FACS-sorterer til >98 % renhet i løpet av 3 4 sorteringer. 20 Figur 2F viser IgE/M1-antistoffenes evne til å binde rhesus-m1 og krabbemakak-m1'. 47H4 og 7A6 binder både rhesus- og krabbemakak-m1', mens 26A11 bare binder rhesus-m1'. 2 I tillegg til de Daudi-transfektante cellestammene vurderte vi binding av IgE/M1'-antistoff til U266, en human IgE-myelomcellestamme (ATCC, Manassas, VA, #TIB196). U266-cellene oppbevares ved høy tetthet (1 2 x 6 /ml) i hybridommedium [RPMI 1640, 1 % kalvefosterserum, 2 mm L- glutamin, mm HEPES, 4, g/l glukose, 1, g/l bikarbonat]. U266-cellene ble farget med 1 µg/ml antistoff mot M. IgE/M1 -antistoffet 47H4 bandt U266 mens 26A11 og 7A6 ikke gjorde det (figur 2E). 30 Eksempel 3B 3 Bindingsspesifisiteten til humanisert antistoff mot IgE/M1' Eksempel 3B viser binding av humaniserte antistoffer mot IgE/M1' til en samling cellestammer som uttrykker IgE/M1 fra menneske, rhesusape eller krabbemakak. Ved å anvende de samme humane B-cellestammene Daudi eller BJAB som overtrykker M1 fra menneske, rhesusape og krabbemakak som omtalt i
136 134 eksempel 3A, ble spesifisiteten til humaniserte kloner av antistoffer mot IgE/M1' testet på den humane B-cellestammen Daudi infisert med retrovirus for å uttrykke den lange formen av IgE med M1'-sekvensen eller den korte formen av IgE som mangler M1'-sekvensen. De humaniserte antistoffene mot IgE/M1' (47H4v, 26A11v6, 7A6v1) er spesifikke for den lange formen og ikke den korte formen (ingen M1') [figur 2G]. HERCEPTIN anti-her2 MAb huigg1 ble anvendt som isotypekontroll (Genentech, Inc). I tillegg binder de humaniserte antistoffene mot IgE/M1' (47H4v og 26A11v6) U266-cellestammene (figur 2H). Figur 2I viser de humaniserte anti-ige/m1-antistoffenes evne til å binde både rhesus- og krabbemakak-m1'. Antistoffene 47H4v og 7A6v1 binder både rhesus- og krabbemakak-m1', mens 26a11v6 bare binder rhesus-m1' Eksempel 4 Relativ bindingsaffinitet for antistoffer mot M1' Vi observerte mange bindingsintensiteter for antistoffer mot IgE/M1' ved 1 µg/ml. For å vurdere den relative affiniteten til disse antistoffene for IgE-reseptoren farget vi BJAB lang-celler med en fortynningsserie av M1'-antistoff (1, 0,1, 0,01, 0,001 µg/ml). BJAB lang-celler ble blokkert med museserum og humant serum ( µl) i 20 minutter på is. Cellene ble farget med passende mengde antistoff mot M1', eller isotypekontrollene anti-gp 120 migg1 eller anti-beiskambrosia migg2a i ytterligere 20 minutter. Cellene ble deretter vasket i FACS-vaskebuffer (1 ml), sentrifugert (1200 rpm i minutter) og deretter resuspendert i 0 µl FACsvaskebuffer med kaprint antimurint IgG-PE-antistoff ( µl) (CALTAG/Invitrogen, Carlsbad, CA #M ) for detektering. Cellene ble vasket på nytt som tidligere og deretter analysert på en FACSCalibur-maskin (BD, Inc., Franklin Lakes, NJ). Spenninginnstillingene bygde på isotypekontrollen med 0,001 µg/ml. Ut fra disse resultatene ble IgE/M1 -antistoffkandidatene rangert etter relativ bindingsaffinitet: 42H4 > 7A6, 47H4, 47G4 > 42A, 26A11, 4C1 > 1D2, 26B11 > 1C11 > 28E9 3 I tillegg til relative affinitetsbestemmelser ved antistofftitrering og FACSdetektering, bestemte vi også affiniteten til valgte M -antistoffer for BJABcellestammer som uttrykker lang form av IgE ved Scatchard-analyse. BJAB lang-celler ble fotberedt for binding i kald bindingsbuffer som besto av basismedium med mm HEPES ph 7,4, og 2 % FBS samt 40 µg/ml humant
137 13 IgG. Cellene ble fortynnet til en konsentrasjon på 1,7 x 6 celler/ml og oppbevart på is inntil de skulle tilsettes til analysen. Antistoffene ble jodert med Iodogen-metoden. Forskjellige konsentrasjoner av umerket antistoff fremstilles i triplikat ved hjelp av en 1:2-fortynning som starter med metningskonsentrasjonen og slutter ved en konsentrasjon på null, med totalt 14 konsentrasjoner. Jodert antistoff med en enkelt konsentrasjon ble tilsatt til alle fortynningene av det umerkede konkurrentantistoffet. Til slutt ble hver blanding av jodert og umerket antistoff tilsatt BJAB lang-celler. Prøvene ble inkubert og ristet ved 4 C i 4 timer. Hver prøve ble så filtrert på en membran, vasket minst 3 ganger med bindingsbuffer, satt til tørking og så målt i 1 minutt med Perkin Elmer Wizard 1470 Auto Gamma Counter. Eksempel 4A Scatchard-affinitet for murine antistoffer mot IgE/M1' BJAB- eller Daudi-cellene som uttrykker forskjellige former for IgE med eller uten M1' (menneske, rhesus, krabbemakak) ble forberedt for binding i kald bindingsbuffer som besto av basismedium med mm HEPES ph 7,4, og 2 % FBS samt 40 µg/ml humant IgG. Cellene ble fortynnet til en konsentrasjon på 1,7 x 6 celler/ml og oppbevart på is inntil de ble tilsatt til analyseblandingen. Proteinene/antistoffene ble jodert ved Iodogen-metoden. Forskjellige konsentrasjoner av kaldt protein fremstilles i triplikat ved hjelp av en 1:2-fortynning som starter med metningskonsentrasjonen og slutter ved en konsentrasjon på null, med totalt 14 konsentrasjoner. Varmt protein av en enkelt konsentrasjon blir tilsatt til alle fortynningene for å konkurrere med det kalde proteinet. Til slutt blir cellene tilsatt til blandingen av de varme og kalde proteinene; det ble brukt celler per prøve. Analysen oppbevares og ristes ved 4 C i 4 timer. Alle prøvene blir så filtrert på membranen, vasket minst 3 ganger med bindingsbuffer, satt til tørking og så telt i 1 minutt med Perkin Elmer Wizard 1470 Auto Gamma Counter. Figur 3O oppsummerer affinitetene til de murine anti-m1'-antistoffene målt ved Scatchard-analyse. Det murine antistoffet 47H4 migg1 bandt M1' fra menneske, rhesusape og krabbemakak med affiniteter på henholdsvis 0,79, 0,77 og 0,97 nm. De murine antistoffene 26A11 migg1 og 7A6 migg1 bandt humant M1' med affiniteter på henholdsvis 2,3 og 0,64 nm.
138 136 Figur 3P oppsummerer bindingsaffinitetene til de humaniserte anti-ige/m1- antistoffene til M1 fra menneske, rhesusape og krabbemakak. Det humaniserte antistoffet 47H4v huigg1 bandt M1 fra menneske, rhesusape og krabbemakak med affiniteter på henholdsvis 1,, 1,3 og 2, nm. De humaniserte antistoffene 47H4 v2, 47H4 v1, 26A11 v6, 26A11 v14 og 26A11 v1 bandt human M1' med affiniteter på henholdsvis 0,4, 0,37, 1,8, 1, og 1,06 nm. Eksempel Bindings-/blokkeringsstudier av Anti-IgE/M1'-epitop Blokkeringsstudiene ble utført mellom kandidatantistoffer mot IgE/M1' for å bestemme overlappende eller distinkte bindingsepitoper. Når de blokkerende og detekterende antistoffene var av forskjellig isotype (dvs. IgG1 vs. IgG2a) ble disse isotypeforskjellene utnyttet for å bestemme graden av blokkering ved å detektere med et isotype-spesifikt sekundært antistoff (CALTAG/Invitrogen, Carlsbad, CA, kaprint antimurint IgG1-PE #32004, eller kaprint antimurint IgG2a #M32204). Når blokkerings- og bindingsantistoffene hadde samme isotype, ble antistoffene konjugert til Biotin (Pierce, Rockford, IL, EZ-link-sulfo-NHS-LC- Biotin #CAS ) og detektert med Streptavidin-PE (BD #4061), eller konjugert til Alexa-647 (Molecular Probes/Invitrogen, Carlsbad, CA, #A20173). Innledningsvis ble blokkerings- og detekteringsantistoffene anvendt i forholdet 1:1 ( µg/ml) (figurene 4A-4B). Antistoffkombinasjoner som innledningsvis ikke ga noen blokkering eller bare delvis blokkering ble testet videre med :1 forhold mellom blokkerings- og detekteringsantistoff. BJAB lang-celler ble blokkert med museserum og humant serum (som beskrevet ovenfor) og deretter farget med blokkeringsantistoffet ( µg/ml) i FACS-vaskebuffer (2 % FBS/1X PBS) i 20 minutter på is. Cellene ble vasket i FACS-vaskebuffer og deretter farget med detekteringsantistoffet i et forhold på 1:1 ( µg/ml) eller :1 (1 µg/ml) i 20 minutter på is. Cellene ble vasket på nytt som ovenfor og deretter analysert på en FACSCalibur-maskin (BD, Inc, Franklin Lakes, NJ). De fargede cellene ble sammenlignet med et isotypekontrollantistoff, enten anti-gp120 migg1 (Genentech, Inc.) eller anti-beiskambrosia-migg2a (Genentech, Inc.) fig. 4C). Til sammenligning ble fullstendig blokkering bestemt ved farging med samme antistoff ukonjugert hvis mulig. Blokkeringsstudiene avdekket de tre store bindingsgruppene A og B og C. Gruppe A ble ytterligere delt inn i to grupper A1 (47H4, 47G4, 42H4, 42A) og
139 137 A2 (4C1, 1D2, 26A11) ut fra på delvis overlappende epitoper (fig. 4D). Gruppe C (26B11) hadde svært liten overlapping med andre antistoffkandidater. Tre kandidater overlappet bredere enn de andre. 1C11 syntes å ha overlappende binding med gruppen A1 og A2, og ble bare delvis blokkert av gruppe B (28E9). 7A6 tilhører primært gruppen A1, men hadde delvis interaksjon med gruppe A2-kandidatene. 28E9 hører til i en gruppe helt for seg selv. 28E9 hadde bare delvis interaksjon med gruppe A2-kandidatene Eksempel A Epitopkartlegging Figur A E viser epitopbindingsstudier utført med antistoffer mot IgE/M1'. De murine antistoffene 47H4, 7A6 og 26A11 er vist i figur B og D mens de humaniserte variantene 47H4v, 7A6v1 og 26A11v6 er vist i figur C og E. Epitopkartleggingsstudier: Murine kandidater Det ble lagd 1 peptider gjennom hele sekvensen som omgir og inkluderer humant M1' (Clifford Quan) (figur A). Peptidene ble resuspendert i enten dh20 eller 0 % DMSO. Peptidene ble belagt på 96-brønners plater (NUNC MaxiSorp ELISA-plater # med høy proteinbindingskapasitet) med 1 µg/ml i beleggingsbuffer (0,0 M karbonat/bikarbonat (ph 9,6) (0 µl per brønn). M1'-Fc-fusjonsproteinet ble anvendt som en positiv bindingskontroll. De negative kontrollene inkluderte brønner belagt med humant IgE (U266) og ikke-belagte brønner. Peptidene ble satt til belegging over natten ved 4 C. Neste morgen ble platene vasket 3 ganger med vaskebuffer (PBS/0,0 % Tween-20 (ph 7,4)). Platene ble deretter blokkert i blokkeringsbuffer (PBS, 0, % BSA (ph 7,4)) i 1 time med forsiktig omrøring. Fortynninger av antistoffene (40 ng/ml og 1 ng/ml) som skulle testes (47H4 migg1, 47H4v huigg1, 26A11 migg1, 26A11v6 huigg1, 7A6 migg1, 7A6v1 huigg1) ble gjort i Magic Buffer [PBS (ph 7,4), 0, % BSA, 0,0 % Tween-20, ppm Proclin., 0,2 % BgG, 0,2 % Chaps, mm EDTA, 0,3 M NaCl]. Antistoff mot gp120 migg1 og HERCEPTIN huigg1-antistoff ble anvendt som kontroller for henholdsvis murine og humane antistoff mot IgE/M1'. Når platene var tilstrekkelig blokkert (0, % BSA/1X PBS), ble de vasket 3 ganger og så ble antistoffene tilsatt til platen i triplikat (0 µl per brønn). Platene ble så inkubert i 2 timer ved romtemperatur. De murine antistoffplatene ble vasket 3 ganger og så tilsatt antimurint IgG1-Biotin (BD Biosciences (A8-1) #3441) (1: 000, 0 µl per brønn) og inkubert i 1 time ved RT. Etter de 3 vaskingene ble brønnene tilsatt SA-HRP (BD Biosciences
140 138 #4066) (1:20 000, 0 µl per brønn) og inkubert i 1 time. De humane antistoffplatene ble vasket som før, tilsatt anti-huigg.fc-hrp (Jackson ImmunoResearch (# )) (1: 000, 0 µl per brønn) og inkubert i 1 time ved RT. Når de sekundære inkuberingene var fullstendige, ble platene vasket 6 ganger og tilsatt TMB-substrat (BD OptEIA #214) (0 µl per brønn). Substrat/HRP-reaksjonen ble stanset med 1 M H 3 PO 4 etter ~4 minutter. Platene ble så avlest ved 40/60. Data ble representert som relativ OD (OD40:CD60). 1 Figur B og D illustrerer binding av murine 47H4, 26A11 og 7A6 anti-ige/m1'- kandidater til M1'-peptider. Binding av disse kandidatene korrelert med epitopgrupperinger definerte antistoffblokkeringsstudiene. 47H4 migg1 bandt peptid 4 (SAQSQRAPDRVLCHS) (SEQ ID NO:8). 7A6 migg1 bandt peptid 4 (SAQSQRAPDRVLCHS) (SEQ ID NO:8) og (RAPDRVLCHSGQQQG) (SEQ ID NO:9). 26A11 migg1 bandt peptid 7 (GQQQGLPRAAGGSVP) (SEQ ID NO:11) og 8 (PRAAGGSVPHPRCH) (SEQ ID NO:12) Figur C og E illustrerer bindingen av de humaniserte antistoffene mot IgE/M1' til M1'-peptidene. Epitopkartleggingen ble utført som beskrevet ovenfor for de tilsvarende murine antistoffene, bortsett fra at det ble anvendt et annet sekundært antistoff. De humane IgG1-antistoffplatene ble vasket som før og tilsatt sekundært antistoff mot IgG.Fc-HRP (Jackson ImmunoResearch (# ) (1: 000, 0 µl per brønn) og inkubert i 1 time ved RT. Epitopbindende paralleller som ble observert med de murine opphavsantistoffene: 47H4 v binder peptid 4 (SAQSQRAPDRVLCHS) (SEQ ID NO:8). 7A6 v1 bandt peptid 4 (SAQSQRAPDRVLCHS) (SEQ ID NO:8) og (RAPDRVLCHSGQQQG) (SEQ ID NO:9). 26A11 migg1 bandt peptid 7 (GQQQGLPRAAGGSVP) (SEQ ID NO:11) og 8 (PRAAGGSVPHPRCH) (SEQ ID NO:12) Eksempel 6 Apoptose utløst av murint anti-ige/m1' i Daudi-IgE/lang-celler Daudi lang (IgE/M1')-celler ble anvendt til å teste virkningen av å kryssbinde overflate-ige på utløsning av apoptose. Daudi-celler (ATCC, Manassas, VA, #CCL-213) er en human Burkitts lymfom-cellestamme som er kjent for å være mottagelig for apoptose som respons på BCR-kryssbinding. Daudi-celler som uttrykker den lange formen for IgE ble produsert ved retroviral transduksjon av de tunge og lette kjedene av U266-IgE med M1', som beskrevet ovenfor for
141 danningen av BJAB IgE lang-celler. Flowcytometrianalyse av Daudi-IgE/langcellene ved hjelp av antistoffer mot endogent IgM og transfektert B- cellereseptor for IgE tyder på høyere uttrykking av IgM enn av IgE. Apoptose av Daudi-IgE/Long-celler ble undersøkt i celler dyrket til en tetthet på 0,2 x 6 /1, ml i dyrkingsmedium [RPMI, % FBS (Hyclone, Logun, UT, #SH ), penicillin:streptomycin (Gibco/Invitrogen, Carlsbad, CA # ), 2 mm glutamin (Genentech, Inc.), mm HEPES (7,2) (Genentech, Inc.), 1 mm Na-pyruvat (Gibco/Invitrogen, Carlsbad, CA #11360), 1,9 g/l natriumbikarbonatløsning (Gibco/Invitrogen, Carlsbad, CA # )]. Før starten av apoptoseanalysen ble de døde cellene fjernet over en ficoll-gradient (GE Healthcare # ) for å redusere bakgrunnsnivået av celledød i analysen. 0,2 x 6 celler ble dyrket i triplikat med og uten antistoffer mot M' eller kontrollantistoffer i løsning i 72 timer. Cellene ble så høstet og analysert på apoptosenivå ved hjelp av Annexin V-FITC Apoptosis Detection Kit I (BD Biosciences, San Jose, CA #647). Cellene ble vasket to ganger i kald PBS (Genentech, Inc.) og deretter resuspendert i 0 µl 1X bindingsbuffer (0,1 M Hepes/NaOH (ph7,4), 1,4 M NaCl, 2 mm CaCl 2 ) (BD Biosciences, San Jose, CA, # E). Så ble cellene farget i mørket med 2, µl av Annexin V-FITC-antistoffet (BD Biosciences, San Jose, CA #1-6874X) og µl propidiumjodid (PI) (BD Biosciences, San Jose, CA # E). Etter 1 minutter ble alle rørene tilsatt 400 µl 1X bindingsbuffer, og cellene ble analysert på en FACSCalibur-flowcytometrimaskin (BD, Inc. Franklin Lakes, NJ). Det ble samlet rundt hendelser for hver prøve. De døende cellene defineres som annexin V-positive og PI-negative. De døde cellene er positive for både annexin V og PI. Andelen av hver populasjon (døde og døende) ble beregnet med FlowJo FACS-analyseprogram (Tree Star, Inc., Ashland, Oregon). Det ble tatt gjennomsnitt av data i triplikat og standardavvikene ble beregnet. Prosent apoptose ble beregnet som summen av de døde og døende cellene og fremstilt grafisk med Excel (Microsoft, Inc.). Kamptotecin (Sigma-Aldrich, St. Louis, MO, #C mg) og antistoff mot IgM (JacksonImmuno Research, West Grove, PA, # ) ble anvendt som positive kontroller for apoptoseutløsning i Daudi- IgE/lang-cellestammen. Antistoffene mot gp120 migg1 eller beiskambrosia-migg2a (Genentech, Inc.) ble anvendt som negative kontroller for denne analysen. Daudi-IgE/langcellene ble dyrket med en rekke konsentrasjoner av antistoff mot IgE/M1 eller isotypekontroll (2,, 1, 0,1, 0,01, 0,001 µg/ml). De testede antistoffene
142 140 mot IgE/M1' var 7A6, 47H4, 47G4, 42A, 42H4, 1C11, 26A11, 1D2, 4C1, 26B11 og 28E9 (Genentech, Inc.). Antistoffet mot humant IgE (Mae11-mIgG1) ble også anvendt for sammenligningsformål. Antistoffene mot gp120 og beiskambrosia utløse ikke mer apoptose enn ubehandlede celler. Bakgrunnsnivået av apoptose lå i området 1 % for hvert forsøk. IgE/M1 -antistoffene 7A6 (> µg/ml), 47H4 (> µg/ml) og 47G4 (2 µg/ml) utløste apoptose av Daudi IgE lang-celler. 26A11 utløste lignende størrelser av apoptose (~30-0 %), men ved mye lavere konsentrasjoner (> 0,1 µg/ml). 42A, 1D2, 4C1, 26B11 og 28E9 utløste ikke apoptose over bakgrunnsnivået. Mae11 utløste heller ikke apoptose over bakgrunnsnivået Vi utførte også apoptoseanalysen i nærvær av et sekundært kaprint antimurint IgG F(ab')2-antistoff (Jackson ImmunoResearch, West Grove, PA # ) for å superkryssbinde IgE-B-cellereseptoren på Daudi-cellestammen. Kryssbindingsforsøkene ble utført i nærvær av kaprint antimurint IgG F(ab')2- antistoff (30 µg) (Jackson ImmunoResearch, West Grove, PA, # ). Med unntak for 26B11 (30 %) utløste alle antistoffene apoptosenivåer på %, men ved forskjellige konsentrasjoner. Disse antistoffene kan klassifiseres i to grupper. 7A6, 1C11, 26A11, 1 D2, 4C1 og 28E9 utløste maksimal apoptose ved de høyeste konsentrasjonene. Den andre gruppen 47H4, 47G4, 42A og 42H4 og MAE11 viste reduserende apoptose ved høyere konsentrasjoner Eksempel 6A Apoptose utløst av humaniserte antistoffer mot IgE/M1' i Daud/IgE lang-celler Daudi lang (IgE/M1')-cellene ble anvendt for å teste virkningen av å kryssbinde overflate-ige på utløsning av apoptose i denne cellestammen. Daudi-celler (ATCC, Manassas, VA, #CCL-213) er en human Burkitt-lymfomcellestamme som er kjent for å være mottagelig for apoptose som respons på BCRkryssbinding. Daudi-IgE/Long-cellene ble dyrket til en tetthet på 0,2 x 6 /1, ml i dyrkingsmedium [RPMI, % FBS (Hyclone, Logun, UT, #SH ), penicillin:streptomycin (Gibco/Invitrogen, Carlsbad, CA # ), 2 mm glutamin (Genentech, Inc.), mm HEPES (7,2) (Genentech, Inc.), 1 mm Na-pyruvat (Gibco/Invitrogen, Carlsbad, CA #11360), 1,9 g/l natriumbikarbonatløsning (Gibco/Invitrogen, Carlsbad, CA # )] natten før og deretter ble de døde cellene fjernet over en ficollgradient
143 (GE Healthcare # ) neste dag. Dette reduserte bakgrunnsnivået av celledød i analysen. 0,2 x 6 celler ble dyrket i triplikat med og uten antistoffer mot M' eller kontrollantistoffer i løsning i 72 timer. Cellene ble så høstet og analysert på apoptosenivå ved hjelp av Annexin V-FITC Apoptosis Detection Kit I (BD Biosciences, San Jose, CA #647). Cellene ble vasket to ganger i kald PBS (Genentech, Inc.) og deretter resuspendert i 0 µl 1X bindingsbuffer (0,1 M Hepes/NaOH (ph 7,4), 1,4 M NaCl, 2 mm CaCl 2 ) (BD Biosciences, San Jose, CA, # E). Så ble cellene farget i mørket med 2, µl av Annexin V-FITC-antistoffet (BD Biosciences, San Jose, CA #1-6874X) og µl propidiumjodid (PI) (BD Biosciences, San Jose, CA # E). Etter 1 minutter ble alle rørene tilsatt 400 µl 1X bindingsbuffer, og cellene ble analysert på en FACSCalibur-flowcytometrimaskin (BD, Inc. Franklin Lakes, NJ). Det ble samlet rundt hendelser for hver prøve. De døende cellene defineres som annexin V-positive og PI-negative. De døde cellene er positive for både annexin V og PI. Andelen av hver populasjon (døde og døende) ble beregnet med FlowJo FACS-analyseprogram (Tree Star, Inc., Ashland, Oregon). Det ble tatt gjennomsnitt av data i triplikat og standardavvikene ble beregnet. Prosent apoptose ble beregnet som summen av de døde og døende cellene og fremstilt grafisk med Excel (Microsoft, Inc.). Et antistoff mot IgR4 (JacksonImmuno Research, West Grove, PA, # ) ble anvendt som en positiv kontroll for utløsning av apoptose i Daudi-IgE/langcellestammen. HERCEPTIN huigg1 (Genentech, Inc.) ble anvendt som en negativ kontroll for denne analysen. Daudi-IgE/lang-cellene ble dyrket med en rekke konsentrasjoner av antistoff mot IgE/M1 eller isotypekontroll (2,, 1, 0,1, 0,01, 0,001 µg/ml). De sekundære kryssbindingsforsøkene ble utført i nærvær av kaprint antimurint IgG F(ab') 2 -antistoff (0 µg) (Biosource #AHI1301). De testede antistoffene mot IgE/M1' var 47H4v, 26A11 v6, 7A6v1 huigg1. 30 Isotypekontrollen HERCEPTIN huigg1 utløste ikke apoptose mer enn ubehandlede celler. Bakgrunnsnivået for apoptose lå i området 1 % for hvert forsøk. Antistoffene 47H4 v, 26A11v6 og 7A6v1 mot IgE/M1' utløste apoptose i området % (figur 8A). 3 Vi gjentok de samme analysene i nærvær av et sekundært kaprint antihumant IgG F(ab )2-antistoff (Biosource #AHI1301) for å kryssbinde IgE-reseptoren på Daudi-cellestammen. Alle antistoffene utløste maksimale apoptosenivåer på
144 %, med en viss nedgang i apoptoseaktiviteten ved høy konsentrasjon av primært antistoff mot IgE/M1' (figur 8B). Villtype vs. afukosylert 47H4v i utløsning av apoptose uten sekundær kryssbinding. En afukosylert (AF) versjon av 47H4 v ble produsert ved hjelp av BIOWAcellestammer (Genentech, Inc.). Både WT- og AF-versjonene av 47H4 v kunne utløse liknende apoptosenivå (figur 8C). Tabell 1 Humaniserte anti-ige/m1'-bindingsaffiniteter Scatchard BJAB-IgE Biacore A0 Biacore 2000 Sekvens- cellebinding M1 M1 IgG forandringer immo- immo- immo- bilisert bilisert bilisert L1- H1- KD IC 0 IC 0 KD KD KD 31/32 34/3 (nm) (nm) (nm) (nm) (nm) (nm) mu26a11 NS MM 1,6 nm 12 2/26 0,3,1 26A11.v1 NS MM ,2 1,7 9,0 26A11.v2 NA MM 18 0,3 26A11.v3 NY MM 22 0,0 2,8 26A11.v4 NS IM ,9 26A11.v NA IM A11.v6 NY IM ,6 8,3 26A11.v7 NS IH 890 meget svak
145 143 Humaniserte anti-ige/m1'-bindingsaffiniteter Scatchard BJAB-IgE Biacore A0 Biacore 2000 Sekvens- cellebinding M1 M1 IgG forandringer immo- immo- immo- bilisert bilisert bilisert L1- H1- KD IC 0 IC 0 KD KD KD 31/32 34/3 (nm) (nm) (nm) (nm) (nm) (nm) 26A11.v NS IN ~ A11.v11 NA IN A11.v12 NY IN A11.v13 SS MM 19 4,0 26A11.v14 QS MM 19 3,9 26A11.v1 SS IM 32 8,4 26A11.v16 QS IM 27 8,8 27e/28 34 L1- H1- H3-0k mu47h4 NG M M 0,4 nm 62 7,6 12 1, 47H4.v1 NG M M ,8 3,1 47H4.v2 NA M M 38 7,4 2,8 47H4.v3 NG I L H4.v4 NA I L > H4.v NA I M H4.v6 NA M L 170 4
146 144 Humaniserte anti-ige/m1'-bindingsaffiniteter Scatchard BJAB-IgE Biacore A0 Biacore 2000 Sekvens- cellebinding M1 M1 IgG forandringer immo- immo- immo- bilisert bilisert bilisert L1- H1- KD IC 0 IC 0 KD KD KD 31/32 34/3 (nm) (nm) (nm) (nm) (nm) (nm) H1-34 H3-0k mu7a6 M M 0,6 nm 2 4, 7A6.v1 M M A6.v2 I L Eksempel 7 1 Utløst kalsiumfluks Vi undersøkte M1'-antistoffenes evne til å utløse kalsiumfluks i Daudi/- IgE/lang-celler som bevis på at disse antistoffene kan utløse cellulær signalering nedstrøms for B-cellereseptoren for IgE. Daudi-celler (ATCC, Manassas, VA, #CCL-213) som overuttrykker den lange formen av IgE/M1' ble dyrket over natten med en lav tetthet på 0,2 x 6 /ml. Neste morgen ble de døde cellene fjernet over en ficollgradient (GE Healthcare # ). Cellene ble lastet med rikelig Indo-1 for å sikre jevn lasting blant alle stimulansene. Cellene ble resuspendert ved x 6 /ml i dyrkingsmedium (RPMI- % FBS (Hyclone), penicillin/streptomycin, 2 mm L-glutamin) og kombinert med kalsiumdetekteringsfargestoffet Indo-1 ( µm) (Molecular probes/invitrogen, Carlsbad, CA, #11223). Så ble cellene vasket i kulturmedium og resuspendert ved 6 /ml. 6 celler ble anvendt per stimulus. Prøvene ble oppvarmet i et 37 C vannbad i minutter før kjøring i en FACs Vantage-maskin (BD, Inc, Franklin Lakes, NJ). Prøvene ble først kjørt i 1
147 14 minutt for å lage en grunnlinje, og deretter ble de stimulert med 20 µg av hvert antistoff, bortsett fra anti-igm ( µg). Data ble samlet inn i 8 minutter. Kinetisk analyse ble utført på FlowJo FACS-analyseprogram (Tree Star, Inc., Ashland, Oregon). Data ble rapportert som forholdet mellom Indo-1 40 bundet/indo fritt. Alle antistoffene mot M1' ble sammenlignet med isotypekontroll anti-gp120 migg1 (20 µg). Anti-humant IgM (Jackson Immuno- Research, West Grove, PA, # ) og anti-ige (Mae11, Genentech, Inc.) ble anvendt som positive kontroller. 7A6, 47H4, 47G4, 26A11 og 1C11 utløste kalsiumfluks i Daudi IgE lang-celler. 28E9 og 42A utløste et lavt nivå av kalsiumfluks. 4C1, 26B11 og 1D2 utløste ikke kalsiumfluks. Eksempel ADCC utløst av anti-ige/m1' 47H4 v wt og afukosylert Antistoffavhengig celleformidlet cytotoksisitet (ADCC) gjør det mulig for cytotoksiske celler å binde seg (gjennom antistoffet) til den antigenbindende målcellen og deretter drepe målcellen med cytotoksiner. Antistoffer mot IgE/M1' som viser ADCC-aktivitet eller økt ADCC-aktivitet kan ha økt terapeutisk verdi ved behandling av IgE-formidlede forstyrrelser. Det har blitt oppdaget at antistoffer produsert i afukosylerte pattedyrceller har økt ADCC-aktivitet. Det følgende forsøket beskrev produksjon av afukosylert antistoff mot IgE/M1' NK-cellene ble isolert fra 0 ml helblod (RosetteSep #6, Stem Cell Technologies). Renheten til NK-cellene ble bestemt ved anti-human CD6- farging. >70 % ren CD6+ NK-celle ble anvendt i hver analyse. Antistoffer mot IgE/M1' og HERCEPTIN anti-her2 MAb huigg1 isotypekontroll ble titrert i serie. Disse antistoffene (0 µl) ble inkubert med BJAB-celler som overuttrykker humant IgE/M1' på celleoverflaten i 30 minutter ved romtemperatur i RPMI-1640 (uten fenolrødt) (BioWhitaker #12-918F) med 1 % FBS (cellestammen ble produsert som beskrevet ovenfor). Cellestammen ble deretter tilsatt NK-celler (0 µl) i et 1:1-forhold ( 000 NK-celler og 000 mål (BJAB-Long)). Analysene ble utført i triplikat. BJAB-huLong, antistoffer og NK-celler ble deretter inkubert i 4 timer ved 37 C. Etter dyrkingen ble 96 brønners U-bunnplater sentrifugert ned og supernatantene ble høstet (0 µl). Supernatantene ble så testet på LDH-frigjøring ved hjelp av LDH-reaksjonsanalysen (Roche # ).
148 146 Målet alene og målet lysert ble anvendt til å beregne den prosentvise cytotoksisiteten. Superantantene ble inkubert ved et likt volum med LDHreaksjonsblandingen som beskrevet av produsenten i minutter. Platene ble deretter avlest ved 490 nm. % cytotoksisitet ble beregnet som følger: = (forventet verdi - mål alene) / mål lysert - mål alene). Data ble plottet ved hjelp av Kaleidagraph og beste tilpasningskurver ble anvendt til å lage ED0-verdier. I figur har isostypekontrollantistoffet HERCEPTIN huigg1 utløst et lavt nivå av cytotoksisitet. Antistoffene mot IgE/M1 utløste spesifikk cytotoksisitet. Villtypeformene og de afukosylerte formene av anti-ige/m1' 47H4v utløste liknende maksimal % cytotoksistet (~70 80 %). Den afukosylerte formen 47H4v var sterkere enn villtypeformen (EC 0 for den afukosylerte formen 47H4v var ~0,83 nm; EC 0 for villtype 47H4v var ~6,6 nm). 1 Eksempel 9 20 Virkningen av murine antistoffer mot IgE/M1' på atopiske hu-scid-mus Figur 12A-I demonstrerer IgE/M1-antistoffenes (både murine og humaniserte) evne til å hemme produksjonen av serum-ige og IgE-produserende plasmaceller i en atopisk hu-scid-modell. I forsøk 1 (# ) ble murine anti-ige/m1'-kandidater testet og i forsøk 2 (# ) ble det testet en humanisert anti-ige/m1'-kandidat. 2 Donorer ble screenet for IgE-konsentrasjoner i serum fra det interne Leukopack-bloddonorprogrammet vårt. To donorer som ble valgt til å gi celler for den atopiske hu-scid-modellen identifiserte seg selv som allergiske og hadde IgE-konsentrasjoner i serum på 1696 og 112 ng/ml. Normale donorer har vanligvis IgE fra <0 til 300 ng/ml Hu-SCID-forsøkene er skissert i figur 12A og 13A. PBMC ble isolert ved leukoforese og ficoll-tetthetsgradient (GE Healthcare # ) og telt. 8 PBMC-celler ( 8 per 0 µl) ble injisert i.p. i bestrålte SCID-beige mus på dag null. For å forskyve responsen mot IgE-produksjon ble musene behandlet med antistoffer mot IFNγ (BD Biosciences, San Jose, CA, #4698) og IL12 (0 µg/dose) (BD Biosciences, San Jose, CA, #469) på dag 0 og 3; og med rhil-4 (0 ng/dose) (R&D Systems, Minneapolis, MN, #204-IL-0) på dag 2,3,4. Musene ble behandlet med forsøksantistoffer (dvs. anti-ige/m1') tre ganger per uke (rundt hver tredje dag) med start på dag 0, inntil slutten av studien. Humane celler ble tillatt å
149 147 ekspandere i mellom 7 og 14 dager og deretter ble musene avlivet. Det ble tatt blodprøver på dag 7 og på slutten av studien. På den siste dagen av forsøket ble musene avlivet og miltcellene ble isolert. Enkeltcellesuspensjoner ble anvendt til IgE Elispot-analyse og FACS for å bestemme de humane cellenes rekonstitueringsgrad og IgE-celledesimeringen i plasma. 1 Dataene ble uttrykt som gjennomsnitt ± standardavvik. P-verdiene ble beregnet med JMP statistikkprogram. I Dunnetts test sammenlignes gruppegjennomsnittene mens alle testgruppene testes mot en referansegruppe. Med en paret Students t-test sammenlignes hvert gruppepar ved hjelp av Students t-test. Så anvendes en Bonferroni-korreksjon til å justere p-verdiene fra den parede Students t-testen for å sikre mot parvise sammenligninger. Alle p-verdierterskelen er 0,0. Prosentvis endring i dataene rapportert i figur 12A-I og 13A-H ble beregnet mellom behandlingsgruppe og kontrollgruppe uten normalisering til grunnlinjekonsentrasjoner Frie IgE-konsentrasjoner ble bestemt med en human ELISA-prosedyre der MaxiSorp 384-brønners ELISA-plater (Nalge Nunc International, Rochester, NY) ble belagt med 1 µg/ml monoklonalt antistoff MAE11 i 0 mm karbonatbuffer, ph 9,6, ved 4 C over natten og blokkert med 0, % bovint serumalbumin i PBS ved romtemperatur i 1 time. Standarder (0,098-12, ng/ml) og tredobbelte seriefortynninger av prøvene (minimum fortynning 1: for å unngå eventuell serumvirkning) i PBS som inneholdt 0, % bovint serumalbumin, 0,0 % polysorbat 20, deler per million Proclin 300 (Supelco, Bellefonte, PA), 0,2 % CHAPS (Sigma, St. Louis, MO), 0,2 % bovine g-globuliner (Biocell, Rancho Dominguez, CA) og mm EDTA ble inkubert på platene ved romtemperatur i 1 time. Bundet IgE ble detektert ved inkubering av peroksidasemerket kaprint antihumant IgE-antistoff (Kirkegaard & Perry Laboratories, Gaithersburg, MD) i brønnene i 1 time. Substratet 3,3',,'-tetrametylbenzidin (Kirkegaard & Perry Laboratories, Gaithersburg, MD) ble tilsatt og reaksjonen ble stoppet ved å tilsette 1 M fosforsyre. Platene ble vasket mellom trinnene. Absorbansen ble avlest ved 40 nm på en Titertek automatleser (ICN, Costa Mesa, CA). Standardkurven ble tilpasset ved hjelp av et kurvetilpasningsprogram med fire-parameters ikke-lineær regresjon (utviklet ved Genentech). Datapunktene som falt i det lineære området av standardkurven ble anvendt til å beregne IgE-konsentrasjonen i prøvene.
150 Isotypepanelet for humant immunoglobulin ble utført ved å ta murint monoklonalt antistoff (mab) mot humant IgG1 (klon 2C11. Abcam Inc., Cambridge, MA) ble fortynnet til 4 µg/ml i 0,0 M natriumkarbonatbuffer, ph 9,6 og belagt på ELISAplater (384-brønner, sterktbindende plater, Greiner Bio One, Monroe, NC) under en inkubering over natten ved 4 C. Etter vasking 3 ganger med vaskebuffer (PBS/0,0 % Tween-20), ble platene blokkert med PBS/0, % bovint serumalbumin (BSA) i 1 til 2 timer. Denne og alle andre inkuberinger ble utført ved romtemperatur på en sirklende rister. Museserumprøvene ble fortynnet 1:0, og så seriefortynnet 1:3 ved hjelp av analysebuffer (PBS/0, % BSA/0,0 % Tween 20/0,2 % CHAPS/ mm EDTA/1PPM Proclin). Ved å anvende den samme bufferen ble det laget til seriefortynninger av IgG1 til standardkurven (12, ng/ml). De forhåndsfortynnede frosne kontrollene for det høye, middels og lave området av standardkurven ble tint. Etter blokkeringstrinnet ble platene vasket, og prøvene, standardene og kontroller ble tilsatt til de 384- brønners platene og inkubert i 2 timer. Platene ble vasket og biotinylert musemab mot humant IgG1 (Zymed Laboratories, South San Francisco, CA) ble fortynnet 1:00 i analysebuffer og tilsatt til de vaskede platene for 1 times inkubering. Streptavidin-pepperrotperoksidase (SA-HRP) (GE Healthcare, Piscataway, NJ) ble fortynnet 1: i analysebuffer og tilsatt til platene etter vaskingen. Etter en 30 min inkubering og et avsluttende vasketrinn ble det tilsatt tetrametylbenzidin (TMB) (Kirkegaard & Perry Laboratories, Gaithersburg, MD) og fargen ble fremkalt i til 1 minutter. Reaksjonen ble stoppet ved å tilsette 1 M fosforsyre. Den optiske tettheten ble målt med en mikroplateleser (40 nm, 60 nm referansebølgelengder), og prøvekonsentrasjonene ble beregnet ut fra 4- parametertilpasninger av standardkurvene. Den minste målbare konsentrasjonen av humant IgG1 i museserumprøvene var 1,22 µg/ml Denne totale ELISA-metoden som beskrives ovenfor ble også anvendt til analysen av de andre Ig-isotypene. IgG2 Murint antihumant IgG2 mab (γ2-kjedespesifikk) (Southern Biotech, Birmingham, AL) ble belagt på ELISA-plater med 4 µg/ml. Standardkurveintervallet for humant IgG2 var 12, ng/ml. Biotinylert murint antihumant IgG2 mab (γ2-kjedespesifikk) (Southern Biotech) ble fortynnet til 0, µg/ml og anvendt som detekteringsantistoff. SA-HRP ble tilsatt til 384-
151 149 brønnersplatene i 1: 000 fortynning. Den minste målbare konsentrasjonen av humant IgG1 i museserumprøvene var 1,22 µg/ml. IgG3 Murint antihumant IgG3 mab (Zymed Laboratories) ble fortynnet til 1 µg/ml og belagt på ELISA-plater. Standardkurveintervallet for humant IgG3 var 0,78 0 ng/ml. Biotinylert murint antihumant IgG3 mab (γ3-kjedespesifikk) (Southern Biotech) ble fortynnet til 0,1 µg/ml og anvendt som detekteringsantistoff. SA-HRP ble tilsatt til 384-brønnersplatene i 1: fortynning. Den minste målbare konsentrasjonen av humant IgG3 i museserumprøvene var 78,1 ng/ml. 1 IgG4 Renset murint antihumant IgG4 mab (BD Pharmingen, San Jose, CA) ble fortynnet til 0,2 µg/ml og belagt på ELISA-plater. Standardkurveintervallet for humant IgG4 var 0,20 2 ng/ml. Biotinylert murint antihumant IgG4 mab (BD Pharmingen) ble fortynnet til 0,2 µg/ml og anvendt som detekteringsantistoff. SA-HRP ble tilsatt til 384-brønners platene i 1: fortynning. Den minste målbare konsentrasjonen av humant IgG4 i museserumprøvene var 39,1 ng/ml IgA Renset murint antihumant IgA 1 /A 2 mab (BD Pharmingen) ble fortynnet til 2 µg/ml og belagt på ELISA-plater. Standardkurveintervallet for humant IgA var 0,20 2 ng/ml. Biotinylert murint antihumant IgA 1 /A 2 mab (BD Pharmingen) ble fortynnet til 1 µg/ml og anvendt som detekteringsantistoff. SA-HRP ble tilsatt til 384-brønners platene i 1: fortynning. Den minste målbare konsentrasjonen av humant IgG4 i museserumprøvene var 39,1 ng/ml IgM Renset murint antihumant IgM mab (BD Pharmingen) ble fortynnet til 0, µg/ml og belagt på ELISA-plater. Standardkurveintervallet for humant IgM var 0,78 0 ng/ml. Biotinkonjugert murint antihumant IgM mab (BD Pharmingen) ble fortynnet til 0, µg/ml og anvendt som detekteringsantistoff. SA-HRP ble tilsatt til 384-brønners platene i 1: fortynning. Den minste målbare konsentrasjonen til humant IgM i museserumprøvene var 16 ng/ml.
152 Hu-SCID-splenocytter, musesplenocyter eller lymfeknuteceller ble telt ved hjelp av Fluoresbrite YG-mikrokuler (Polysciences, Inc. #18862) på et FACSCalibur flowcytometer (BD (San Jose, CA)) Elispot-analyser ble anvendt til å bestemme hyppigheten av IgE-produserende plasmaceller i hu-scid-mus etter behandling med rhuil-4 og antistoffer mot IFNγ og IL-12 i 1 dager. Elispot-plater (Millipore, Billerica, MA, #MSIPS4, klar) ble belagt med en 1:00 fortynning av primært ukonjugert antistoff mot humant IgE (AXXORA, San Diego, CA, #BET-A80-8A) i beleggingsbuffer [Genentech, Inc. (A3323)] med 0 µl/brønn over natten ved 4 C. Deretter ble den primære antistoffløsningen dekantert og platene ble vasket to ganger med vaskebuffer (1X PBS/0,0 % Tween-20). Membranene ble blokkert med 200 µl/brønn ved hjelp av 0, % BSA i 1 X PBS i 0, 1 time ved 37 C. Blokkeringsløsningen ble fjernet og platen ble tilsatt positive kontrollceller eller hu-scid-splenocytter. Den humane myelom-ige-produserende U266-cellestammen (ATCC, Manassas, VA, #TIB196) ble anvendt som positiv kontroll for analysen. U266-cellene ble tilsatt til platen med startkonsentrasjon på 3000 celler/0,1 ml (=0,032 x 6 /ml) hybridommedium [RPMI 1640, 1 % kalvefosterserum, 2 mm L-glutamin, mm HEPES, 4, g/l glukose, 1, g/l bikarbonat], og deretter ble det utført sju seriefortynninger (1:2). Enkeltcellesuspensjonene av hele miltceller ble resuspendert i 1 ml medium. 0 µl ble tilsatt til den første brønnen. Deretter ble det utført sju 1:2 seriefortynninger. U266 hybridom- og miltceller ble inkubert over natten ved 37 C. Neste dag ble platene vasket tre ganger med vaskebuffer (1XPBS, 0,0 % Tween-20) og deretter ble 0 µl/brønn av det sekundære antistoffet, anti-humant IgE alkalisk fosfatasekonjugert (Sigma, St. Louis, MO, A-32) tilsatt med 1:2000 i 0, % BSA/1X PBS til hver brønn og inkubert ved 37 C i 2 timer. Platene ble vasket tre ganger, og så skylt én gang med ddh20 (Genentech, Inc.) Hver brønn ble tilsatt BCIP/NBT-løsning med 0 µl/brønn (R&D Systems, Minneapolis, MN, Elispot Blue Color Module, #SEL002) og inkubert i mørket i 30 minutter. Deretter ble platene skylt med ddh20 én gang og platen ble satt til tørking ved romtemperatur. Platene ble sendt til BD Biosciences for telling (BD, Inc., Franklin Lakes, NJ). Antallet plasmaceller ble beregnet ved å anvende antallet detekterte flekker, antall gangers fortynning (celler tilført) og det totale miltcelleantallet. 3 De humane plasmacellene ble fremstilt ved å fjerne miltene fra antistoffbehandlede hu-scid-mus på dag 1. Enkeltcellesuspensjoner ble fremstilt og filtrert. x 6 celler ble blokkert med humant serum (Sigma, St.
153 11 Louis, MO, #S-227) og museserum (VWR, #RLD8-00), og så farget med anti-humant CD38 PE (BD Biosciences, San Jose, CA, #460) og antistoffer mot humant PC-FITC (Dakocytomation, Glostrup, Danmark, #K2311) in FACsvaskebuffer (2 % FBS, 1X PBS) på is i 20 minutter. Cellene ble så vasket og analysert på en FACSCalibur-maskin (BD, Inc., Franklin Lakes, NJ). Human CD38-uttrykking ble anvendt som en positiv kontroll for vellykket human PBMC-overføring. Plasmacellene ble definert som CD38 høy, PC+. 1 I forsøk # (figur 12A) ble tre antistoffer mot IgE/M1' (47H4, 26A11 og 7A6) testet mot isotypekontrollen anti-gp120-muigg1. Isotypekontrollbehandlede mus dannet høye konsentrasjoner av humant IgE innen dag 9. Behandling med anti- IgE/M1'-kandidater reduserte IgE-konsentrasjonene med 6 84 % (figur 12 B C). Anti-IgE/M1'-behandling reduserte også antall IgE-produserende celler in vivo med % (figur 12D E). Andre serum-ig var relativt upåvirket av anti-ige/m1'- behandling (reduksjoner på ~30 % ble observert med noen M -antistoffer for huig1, IgG3 og IgG4 (figur 12F G). Ingen reduksjon i prosentandelen av de totale plasmacellene til milten ble observert med anti-ige/m1'-behandling (figur 12H I) I forsøk # (figur 13A) ble et humanisert anti-ige/m1' (47H4v) testet mot isotypekontrollantistoffet HERCEPTIN -huigg1. Isotypekontrollbehandlede mus dannet høye serumkonsentrasjoner av humant IgE på dag 8. Behandling med anti-ige/m1' huigg1 reduserte IgE-konsentrasjonene med 79 % (figur 13B D) og IgE-produserende plasmaceller med 7 % (figur 13C D). Ingen reduksjon i andre serum-igg (IgG1, IgG2, IgG3, IgG4, IgM, IgA) (figur 13E F) eller i prosentandelen av de totale plasmacellene i milten ble observert (figur 13G H). Disse studiene viser at antistoffer mot IgE/M1' spesifikt reduserer serum-ige og antallet IgE-produserende celler, men ikke immunoglobuliner av andre isotyper. 30 Eksempel Humane M1'-transgene mus 3 Produksjon av hum1 -transgene «knockin»-mus Siden mus normalt ikke uttrykker et M1'-domene som likner menneskets, produserte vi mus med det humane M1'-domenet «slått inn» i muse-igelokusen (figur 14A). Den humane M1'-sekvensen anvender det murine M1- spleiseakseptorsetet oppstrøms og fusjoneres deretter til den murine M1-
154 12 sekvensen nedstrøms (figur 14B). En IRES-EGFPpA - Neo-kassett ble også «slått inn» i 3' på den murine M2-sekvensen. Denne «innslåingen» gjør det mulig å uttrykke murint IgE med det humane M1'-domenet på overflaten av IgE+ B-celler. M1'-sekvensen uttrykkes ikke på utskilt IgE. Screening av M1 -«knockin»-stammer (PCR) Humane M1 -«knockin»-mus ble genotypet ved PCR med primere spesifikke for den murine IgE-lokusen og den humane M1 -sekvensen. De anvendte primerne var som følger: WT FOR '-GGCCAAAGACCCTAAGACAGTC (SEQ ID NO:6) hum1' FOR '-GGGCTGGCTGGCGGCTCCGC (SEQ ID NO:7) WT REV '-CTATGCCCTGGTCTGGAAGATG (SEQ ID NO:8) 1 Renset genomisk DNA ble analysert med de ovennevnte primerne med 32 sykluser av det følgende programmet [94 C i 4 min; 94 C i 1 min; 60 C i 30 sek; 72 C i 1 min (30 sykluser); 72 C i min]. PCR-produktene ble deretter lagt ut på en 2 % agarose 0, x TBE gel. Villtypegenotyping med PCR ga et 668 bp produkt mens hu-m1 -«knockin» ga et 47 bp produkt (figur 14C) Screening av M1 -«knockin»-stammer (Southern) hum1' «knockin»-es-celler og endelige musestammer ble screenet og verifisert med southern blot. µg renset ES-celle-DNA eller halegenomisk DNA ble digerert med Hindlll for venstre arm eller BamHI for høyre arm over natten. Det digererte DNA ble så lagt ut på en 0,8 % agarose 1X TAE-gel. DNA ble så overført til en nylonmembran (Roche # ) og denatureringsbuffer (1, M NaCl; 0, M NaOH) ble brukt over natten. Membranen ble skylt, UV-kryssbundet, og deretter oppbevart i DIG Easy Hyb-løsning (Roche #18762) i 4 timer med rotasjon ved 46 C. Det ble brukt PCR til å lage prober ved hjelp av PCR DIG probesyntesesett etter produsentens anvisninger (Roche # ). Primerne for den venstre armen var: FOR '-TGTCTGGTGGTGGACCTGGAAAGCG (SEQ ID NO:9) Rev '-TCCTCGCTCTCCTGCTCTGGTGGTG (SEQ ID NO:1) 3 Primerne for den høyre armen var: FOR '-CCATGCAACCTAGTATCCTATTCTC (SEQ ID NO:111) Rev '-CTTTATACAGGAGAACCTAGCCCAG (SEQ ID NO:112)
155 13 Probene ble testet mot umerket PCR-produkt for å sikre økt størrelse og god DIG-merking. Blottene ble deretter probet over natten med kokt probe over natten ved 46 C med rotasjon. Neste dag ble blottene vasket og fremkalt med antistoff mot DIG etter produsentens retningslinjer. Blottene ble eksponert på film i 1 20 minutter. Figur 14D illustrerer southern-resultatene. Villtypemusene danner et 7,4 kb HindIII venstrearmsfragment som blir et 3 kb fragment i hum1 -«knockin»-allelet. Villtypemusene danner et 14,1 kb BamHI høyrearmsfragment som blir et 18,1 kb fragment i hum1 -«knockin»-allelet. Både villtype og heterozygote mus er vist (figur 14D). Eksempel Transgen modell av hu-m1': TNP-OVA-immunisering Dette eksempelet viser anti-ige/m1'-kandidatenes evne til å forhindre produksjon av IgE som skyldes stimulering fra TNP-OVA i både en primær immunrespons (forsøk F, figur 1A I) eller minnerespons (forsøk B, figur 16A K) immunrespons mot TNP-OVA. TNP-OVA eller trinitrofenyl-ovalbumin er et meget sterkt immunogen som ofte anvendes til å lage en sterk antistoffimmunrespons. Dataene ble uttrykt som gjennomsnitt ± standardavvik. P-verdiene ble beregnet med JMP statistikkprogram. I Dunnetts test sammenlignes gruppegjennomsnittene mens alle testgruppene testes mot en referansegruppe. Med en paret Students t- test sammenlignes hvert gruppepar ved hjelp av Students t-test. Så anvendes en Bonferroni-korreksjon til å justere p-verdiene fra den parede Students t-testen for å sikre mot parvise sammenligninger. Alle p-verdier-terskelen er 0,0. Prosentvis forandring i dataene som er rapportert for den primære responsen (figur 1A-I) og minneimmunresponsen (figur 1A K) ble beregnet ved å trekke den ikke-infiserte eller ikke-immuniserte gruppegjennomsnittsverdien fra hver av museverdiene; gruppegjennomsnittene ble beregnet på nytt og den prosentvise endringen ble deretter beregnet mot kontrollgruppen for hvert tidspunkt. 3 TNP-OVA-immunisering av humant IgE/M1 -«knockin»-mus (C7BL/6) utløser en balansert Th1:Th2-respons in vivo. Den antigenspesifikke IgE-konsentrasjonen etter primærimmunisering og utfordring kommer opp i ~ 200 ng/ml. I forsøk F, ble hum1 -«knockin»-mus (C7B1/6) immunisert med TNP-OVA (Biosearch Technologies, Novato, CA) (0 µg i 2 mg alun per mus i.p.) på dag 0
156 14 (figur 1A). Musene ble så behandlet med 0,1 mg/kg antistoff tre ganger per uke mellom dag 0 og 28 (figur 1A). I forsøk F ble hum1 -«knockin»- musene (C7B1:6) immunisert med TNP-OVA/alun på dag 0 (som før) og deretter utfordret med TNP-OVA (0 µg per mus i.p.) på dag 28 (figur 16A). Musene ble så behandlet med mg/kg antistoff mellom dag 28 og 49 (figur 16A). Det ble tatt blodprøver av musene i løpet av immunresponsen for å overvåke antigenspesifikke serumkonsentrasjoner av IgE og IgG TNP-OVA- spesifikk IgE og IgG1 ble målt ved hjelp av ELISA-plater (384-brønners med MaxiSorp -overflate, Nunc, Neptune, NJ) belagt i timer ved 2 8 C med 2 µl TNP-OVA/brønn (TNP:OVA-forhold på 13:1) (Biosearch Technologies, Novato, CA) fortynnet til 0, µg/ml i 0 mm natriumkarbonat/bikarbonat, ph 9,6. Platene ble deretter dekantert og blottet tørre. Platene ble tilsatt blokkeringsbuffer (PBS, 0, % BSA, ph 7,2, 0 µl/brønn) som fikk ligge i 1 2 timer. Denne og alle påfølgende inkuberinger ble utført ved romtemperatur med forsiktig omrøring. Prøvene ble fortynnet 1:0 i analysefortynningsmiddel (PBS, 0, % BSA, 0,0 % Tween-20, 0,01 % Proclin 300) og så seriefortynnet 1:3 gjennom elleve punkter. Standardene ble seriefortynnet dobbelt i analysefortynningsmiddel gjennom sju punkter, med start på 1 ng/ml for anti-tnp OVA IgG1 og 3 ng/ml for anti-tnp OVA IgE. Platene ble vasket tre ganger med vaskebuffer (PBS, 0,0 % Tween-20, ph 7,2) og tilsatt standarder, prøver og kontroller (2 µl/brønn) for å duplisere platene for separat detektering av IgG1- og IgE-isotypen. Etter 1, 2 timers inkubering ble platene vasket seks ganger med vaskebuffer. Biotinylert antimurint IgG1 og IgE mab fra rotter (BD Biosciences, San Diego, CA) ble fortynnet til 0, µg/ml for IgG1 eller 1,0 µg/ml for IgE i analysefortynningsmiddel og tilsatt til de tilsvarende platene (2 µl/brønn). Platene ble inkubert i 1 1, timer og vasket seks ganger med vaskebuffer. Streptavidin/pepperrotperoksidase-konjugat (AMDEX, GE Healthcare, Piscataway, NJ) ble fortynnet 1: for IgG1 og 1: 000 for IgE og tilsatt til platene (2 µl/brønn) hvor det fikk ligge i 30 min. Etter vasking seks ganger med vaskebuffer ble platene fremkalt ved å tilsette 2 µl tetrametylbenzidin/brønn (Kirkegaard & Perry Laboratories, Gaithersburg, MD) og inkubert i 1 min for IgG1 og 2 min for IgE. Reaksjonen ble stanset ved å tilsette 2 µl 1M H 3 PO 4 /brønn, og absorbansen ble avlest ved 40 nm med en 620 nm referanse. Resultatene for ukjente prøver ble interpolert ved 4-parameterstilpasninger til standardkurvene. Museplasmacellene fra milten eller lymfeknutene ble først identifisert ved å separere CD138+-celler. Disse IgE-produserende plasmacellene ble kvantifisert
157 ved Elispot-fremgangsmåten. For alle IgE-produserende celler ble et antistoff mot IgE (fra BD OptEIA mige-settet #248) belagt på Multiscreen-HTSplater (Millipore #MS1PS4W) med fortynning 1:2 og 0 µl ELISAbeleggingsbuffer per brønn. Beleggingen ble utført ved 4 C over natten. Neste morgen ble platene vasket ganger med 200 µl steril PBS-Tween 20 (0,0 %) ved hjelp av en flerkanalspipette i vevskulturavtrekket. Platene ble så blokkert i 300 µl PBS: % BSA eller celledyrkingsmedium (RPMI-1640, % FBS) i to timer ved romtemperatur. Enkeltcellesuspensjonene ble fremstilt fra milt eller lymfeknuter og cellene ble telt ved FACS (som beskrevet ovenfor). Cellene ble klargjort for platelegging, med utgangspunkt på 7 celler og så av 3 4 gangers fortynning for TNP-OVA-immuniseringsmodellene og 4 x 6 celler og så 2 gangers fortynninger for nippostrongylusinfeksjonsmodellene. Den murine A20 B-cellestammen ble anvendt som negativ kontroll og TIB-141- cellestammen ble anvendt som positiv kontroll. Cellefortynningene ble fremstilt i cellekulturmedium (RPMI % FBS). Etter blokkeringstrinnet ble platene vasket én gang med cellekulturmedium og aspirert. Cellene ble lagt på platene med 0 µl per brønn. Platene ble så inkubert over natten ved 37 C, % CO2. Neste dag ble platene vasket 6 ganger med en SkanWasher 300 (Molecular Devices (Sunnyvale, CA)) og PBS-Tween 20 (0,0 %). Så ble det anvendt et biotinylert sekundært antistoff fra BD OptEIA mige-settet (#248) i konsentrasjonen som var angitt av produsenten, vanligvis mellom 1:20 og 1:00 ved 0 µl/brønn. Det sekundære antistoffet ble fremstilt i PBS/1 % BSA. Platene ble så inkubert i 1 time ved romtemperatur og deretter vasket 6 ganger med en SkanWasher300 og PBS-tween 20 (0,0 %). Deretter ble det tilsatt streptavidin AP (R&D #SEL002) i en fortynning på 1:60 med 0 µl per brønn, igjen fremstilt i PBS/1 % BSA og inkubert i 1 time ved romtemperatur. Platene ble vasket som tidligere 3 ganger og så skylt to ganger med ionebyttet vann. Eventuelle væskerester ble tørket av med et papirhåndkle. Så ble fremkallingsreagensen BCIP:NBT (0 µl/brønn) tilsatt, og platene ble inkubert i 30 minutter i mørket ved romtemperatur. Deretter ble substratet dekantert og platene skylt to ganger med ionebyttet vann. Platene ble snudd opp ned for å fjerne overflødig vann og satt til tørking. Flekkene ble kvantifisert ved hjelp av et dissekteringsmikroskop. 3 Forsøket # F testet anti-ige/m1' sin evne til å forhindre en IgE-respons på OVA-TNP-primær immunisering. Mus behandlet med anti-gp120- isotypekontroll produserte serum-ige, og nådde toppkonsentrasjonene mellom
158 16 dag 8 og 14 (~2 ng/ml) (figur 1B). Anti-IgE/M1'-behandlingen hindret økningen i serum-ige på dag 8 (98 %) og 14 (99 %) (figur 1C D). Denne reduksjonen var signifikant forskjellig fra isotypebehandlede dyr og ikke signifikant forskjellig fra ikke-immuniserte mus (figur 1C E). Konsentrasjonene av antigenspesifikk IgG1 ble bare beskjedent påvirket av anti-ige/m1' i løpet av de 28 dagene av forsøket (figur 1F I) I forsøk F testet man evnen hos anti-ige/m1' til å hindre en sekundær/minne-ige-respons på TNP-OVA. Den sekundære IgE-responsen på TNP- OVA-økningen på dag 28 er hurtigere, med en topp etter 4 dager i stedet for 8 9 dager i den primære responsen (figur 16B). Behandlingen med anti-ige/m1'- kandidatene som startet med økning på dag 28, reduserte økningen i IgE mellom dag 28 og 3 (figur 16B). De anti-ige/m1'-behandlede IgE-konsentrasjonene ble signifikant redusert sammenlignet med isotypekontrollen, 9 7 % på dag 32 og % på dag 3 (figur 16C D). På dag 3 42 var IgE-konsentrasjonene ikke signifikant forskjellige fra ikke-immuniserte mus (figur 16E). Analysen av arealet under kurven for disse behandlingsgruppene mellom dag 28 og 49 viser også at anti-ige/m1' reduserte IgE-konsentrasjonene i serum med %, og den gjennomsnittlige daglige konsentrasjonen av antigenspesifikk IgE ble også redusert med % (figur 16F H). Det ble ikke observert noen signifikant reduksjon i antigenspesifikk IgG1 etter behandling med anti-ige/m1' (figur 16I K). Eksempel Hu-M1'-transgen modell: Smittemodellen Nippostrongylus brasiliensis Dette eksempelet illustrerer evnen hos antistoffene mot IgE/M1 til å hindre produksjon av IgE (figur 17A D) og terapeutisk redusere produksjonen av IgE i en tidligere påført Nippostrongylus brasiliensis-infeksjon. Reduksjonen av IgEproduksjonen ble undersøkt ved å administrere antistoffer mot IgE/M1' både på IgE-konsentrasjonstoppen i immunresponsen (figur 18A I), og når de ble anvendt i den sene immunresponsen (figur 19 A G). 3 Nippostrongylus brasiliensis er en gastrointestinal nematode som infiserer rotter og mus. Eggene klekkes i avføringen og modnes til det smittefarlige larvestadiet L3. L3-larvene kommer inn i verten gjennom huden, migrerer til blodårene og går så til lungene etter én til to dager. Etter å ha utviklet seg til L4-larvestadiet i lungene går de langs luftrøret og spiserøret til verten og når tynntarmen på dag
159 17 2 til 3. Larvene modnes til voksne ormer og parer seg og produserer egg rundt dag. N. brasiliensis-egg skilles ut av vertene sammen med avføringen. Mus infisert med N. brasiliensis viser en innledende medfødt immunrespons, og så en sterk type 2-immunitet for å fjerne infeksjonen. I det tidlige stadiet av infeksjonen er det funnet komplement og fibronektin avsatt for å lette rekruttering og tilklebing av leukocytter. Effektorceller, som eosinofiler, basofiler og CD4+Th2-celler, blir rekruttert til lungene for å bekjempe infeksjonen. Th2- cytokinene, IL-4 og IL-13, spiller en avgjørende rolle for å opprettholde immunresponsen i lungene, og er nødvendig for å skille ut ormene i tarmen. Type 2-responsen er karakterisert ved høy antistoffproduksjon, inkludert IgE Humant IgE/M1 -«knockin»-mus (G7BL/6) får en sterk IgE-respons på N. brasiliensis-infeksjon med topp på dag 1 20 (figur 17B, 18B, 19B). IgEkonsentrasjonene i serum faller deretter til en lavere, men vedvarende forhøyet konsentrasjon. Musene ble smittet med Nippostrongylus brasiliensis på dag 0. Alle tre N. brasiliensis-smitteforsøkene ble behandlet med mg/kg antistoffer mot IgE/M1 migg1 eller anti-gp120 migg1-isotypekontroll. I den preventive studien (figur 17A) ble hum1 -«knockin»-musene behandlet tre ganger per uke med start på dag 0 og til og med dag 21. I topproduksjonsstudien (figur 18A) ble hum1 -«knockin»-musene behandlet tre ganger per uke mellom dag 11 og 21. I den sene intervensjonsstudien (figur 19A) ble hum1'-«knockin»-musene behandlet tre ganger per uke mellom dag 41 og Dataene ble uttrykt som gjennomsnitt ± standardavvik. P-verdiene ble beregnet med JMP statistikkprogram. I Dunnetts test sammenlignes gruppegjennomsnittene mens alle testgruppene testes mot en referansegruppe. Med en paret Students t-test sammenlignes hvert gruppepar ved hjelp av Students t-test. Så anvendes en Bonferroni-korreksjon til å justere p-verdiene fra den parede Students t-testen for å sikre mot parvise sammenligninger. Alle p-verdier-terskelen er 0,0. Prosentvis forandring i dataene som er rapportert for den preventive studien og topproduksjonsstudien ble beregnet ved å trekke den ikke-infiserte eller ikke-immuniserte gruppegjennomsnittsverdien fra hver av museverdiene; gruppegjennomsnittene ble beregnet på nytt og den prosentvise endringen ble deretter beregnet mot kontrollgruppen for hvert tidspunkt. I den sene intervensjonsstudien ble prosentvis endring beregnet innenfor hver behandlingsgruppe ved å sammenligne dag 48 og med dag 41.
160 18 De IgE-produserende plasmacellene ble definert og evaluert med Elispot som beskrevet tidligere for eksempel 12. I den preventive studien ble evnen hos anti-ige/m1' til å forhindre økningen i IgE som respons på en primær Nippostrongylus-infeksjon. Kontrollmus (isotypebehandlede) kom opp i høy IgE-produksjon på dag 1 etter infeksjonen (~3000 ng/ml). Anti-IgE/M1'-behandlede mus hadde en lavere IgE-produksjon etter infeksjonen (redusert med % i forhold til anti-gp120 migg1) som ikke var signifikant forskjellig fra produksjonen til ikke-infiserte mus (figur 17B-D) I topproduksjonsstudien ble evnen hos anti-ige/m1' til å redusere IgEkonsentrasjonene terapeutisk testet ved toppen av IgE-responsen på N. brasiliensis-infeksjonen. Alle behandlingsgruppene kom opp i høye IgEkonsentrasjoner (~2000 ng/ml) dag 11 etter Nippostrongylus-infeksjonen (figur 18B). Anti-IgE/M1'-behandlingen startet på dag 11 på toppen av denne responsen. Anti-IgE/M1'-mIgG1-kandidatene reduserte serumkonsentrasjonene av IgE med % innen fire dagers behandling (figur 18C-D). På dag 21 var IgEkonsentrasjonen i de anti-ige/m1'-behandlede musene redusert med % og kom ned i konsentrasjoner som ikke var statistisk signifikante fra den ikke-infiserte kontrollgruppen (figur 18E). De IgE-produserende plasmacellene ble kvantifisert av anti-ige Elispot. N. brasiliensis-infeksjon utløste et betydelig antall IgEproduserende celler både i den mesenteriske lymfeknuten (~30 celler per 4 x 6 ; figur 18F) og i milten (~ celler per 4 x 6 ; figur 18G). På dag 21 reduserte anti- IgE/M1'-behandlingen IgE-produserende celler i den mesenteriske lymfeknuten med % og i milten med 7-66 %. Frekvensen at de totale plasmacellene (CD138+ celler) i de mesenteriske lymfeknutene og milten økte i alle behandlingsgruppene sammenlignet med de ikke-infiserte musene det var ingen signifikant endring i total plasmacellefrekvens i noen av organene som følge av behandling med anti-ige/m1' (figur 18H-I). Disse resultatene demonstrerer evnen hos antistoffene mot IgE/M1 til å redusere IgE-konsentrasjonen i serum ved å desimere de IgE-produserende cellene in vivo, selv når de anvendes på toppnivået av IgE-produksjonen. 3 I den sene intervensjonsstudien ble evnen hos anti-ige/m1' til å redusere den vedvarende lave konsentrasjonen av IgE som nås sent i infeksjonssyklusen. Alle behandlingsgruppene nådde toppen av IgE-produksjonen rundt dag 1. Etter starten av anti-ige/m1'-behandlingen på dag 41 var det noen forskjeller i
161 19 1 serumkonsentrasjonene av IgE blant behandlingsgruppene (figur 19B C), som ble normalisert ved å definere konsentrasjonene på dag 41 som 0 % (figur 19D). Anti-IgE/M1'-behandlingen reduserte serum-ige-konsentrasjonene signifikant sammenlignet med isotypekontrollen anti-gp120 migg1 mellom dag 48 og (figur 19E G). På dag 48 og ble IgE-konsentrasjonene redusert med henholdsvis 64 7 % og 7 84 %, sammenlignet med isotypekontrollen (antigp120 migg1) som ble redusert med henholdsvis 20 % og 37 % (figur 19F G). Forskjellen i gjennomsnittlig IgE-konsentrasjon mellom behandlingsgruppene var ikke signifikant forskjellig fra isotypekontrollgruppen (gp120) før starten av anti- IgE/M1'-behandlingen (figur 19E). Behandling med antistoffer mot IgE/M1' viste imidlertid en mer dramatisk og signifikant reduksjon i serumkonsentrasjonen av IgE enn det som ble observert i kontrollgruppen på dag 48 (figur 19F) og dag (figur 19G). Prosentvis endring og p-verdi (d41) ble beregnet ved å sammenligne hver behandlingsgruppe på dag 48 eller med den samme gruppens startverdi på dag 41, før behandlingen. Eksempel Uttrykking av antistoff mot IgE/M1' i pattedyrceller Dette eksempelet illustrerer fremstilling av potensielt glykosylerte former av det ønskede proteinet eller antistoffet ved rekombinant uttrykking i pattedyrceller. 2 Vektoren, prk (se EP 307,247, publisert 1. mars 1989) benyttes som uttrykkingsvektor. Alternativt bindes DNA som koder for den lette og/eller tunge kjeden til antistoffet mot IgE/M1' i prk med utvalgte restriksjonsenzymer for å gjøre det mulig å sette inn et slikt DNA med slike bindingsmetoder, som beskrives i Sambrook et al., ovenfor I en utførelsesform kan de valgte vertscellene være 293-celler. Humane 293-celler (ATCC CCL 173) ble dyrket til konfluens på vevskulturplater i medium som f.eks. DMEM supplert med kalvefosterserum og eventuelt næringskomponenter og/eller antibiotika. Rundt µg hvis DNA som koder for antistoffet mot IgE/M1' som bindes i prk blandes med rundt 1 µg DNA som koder for VA-RNA-genet [Thimmappaya et al., Cell, 31:43 (1982)] og løses i 00 µl av 1 mm Tris-HCl, 0,1 mm EDTA, 0,227 M CaCl 2. Denne blandingen ble dråpevis tilsatt 00 µl 0 mm HEPES (ph 7,3), 280 mm NaCl, 1, mm NaPO 4 og den ble satt til felling i minutter ved 2 C. Bunnfallet ble suspendert og tilsatt til 293-cellene satt til sedimentering i omtrent
162 160 fire timer ved 37 C. Kulturmediet ble aspirert og 2 ml 20 % glyserol i PBS ble tilsatt i løpet av 30 sekunder. Så ble 293-cellene vasket med serumfritt medium, tilsatt friskt medium og cellene ble inkubert i rundt dager. Omtrent 24 timer etter transfekteringene ble kulturmediet fjernet og erstattet med dyrkingsmedium (alene) eller dyrkingsmedium som inneholdt µci/ml 3Scystein og 200 µci/ml 3S-metionin. Etter en timers inkubering ble det kondisjonerte mediet tatt vare på, konsentrert på et sentrifugeringsfilter og lagt på en 1 % SDS-gel. Den behandlede gelen kan tørkes og eksponeres på film i en valgt tidsperiode for å avdekke nærvær av antistoffet mot IgE/M1'. Kulturene som inneholdt de transfekterte cellene kan gjennomgå ytterligere inkubering (i serumfritt medium), og mediet ble testet i valgt bioanalyse I en alternativ metode kan antistoffet mot IgE/M1' innføres i 293-celler ved kortvarig anvendelse av dekstransulfatmetoden som er beskrevet av Somparyrac et al., Proc. Natl. Acad. Sci., 12:77 (1981). 293-cellene dyrkes til maksimal tetthet i en sentrifugeringskolbe og tilsettes 700 µg DNA som koder for antistoffet mot IgE/M1' som er bundet i prk. Cellene konsentreres først fra sentrifugekolben ved sentrifugering og vaskes med PBS. DNA-dekstranbunnfallet inkuberes på cellepelleten i fire timer. Cellene behandles med 20 % glyserol i 90 sekunder, vaskes med vevskulturmedium og føres tilbake til sentrifugekolben som inneholdt vevskulturmedium, µg/ml bovint insulin og 0,1 µg/ml bovint transferrin. Etter rundt fire dager sentrifugeres og filtreres det kondisjonerte mediet for å fjerne celler og rester. Prøven som inneholder det uttrykte antistoffet mot IgE/M1' kan så konsentreres og renses ved en hvilken som helst valgt fremgangsmåte, som f.eks. dialyse og/eller kolonnekromatografi I en annen utførelsesform kan antistoffet mot IgE/M1' uttrykkes i CHO-celler. DNA-et som koder for antistoffet mot IgE/M1' som er bundet i prk kan transfekteres inn i CHO-celler ved hjelp av kjente reagenser som f.eks. CaPO 4 eller DEAE-dekstran. Som beskrevet ovenfor kan cellekulturene inkuberes, og mediet erstattes med dyrkingsmedium (alene) eller i medium som inneholder en radiomerking som f.eks. 3 S-metionin. Etter at nærværet av antistoffet mot IgE/M1' er bestemt, kan dyrkingsmediet erstattes med serumfritt medium. Fortrinnsvis inkuberes kulturene i omtrent 6 dager, og deretter høstes det kondisjonerte mediet. Mediet som inneholder det uttrykte antistoffet mot IgE/M1' kan så konsentreres og renses ved en hvilken som helst valgt fremgangsmåte.
163 161 Epitopmerkede varianter av antistoffet mot IgE/M1' kan også uttrykkes i CHOvertsceller. DNA som koder for antistoffet mot IgE/M1' som er bundet i prk, kan subklones ut av prk-vektoren. Ved hjelp av PCR kan den subklonede innsatte sekvensen fusjoneres i ramme med en valgt epitopmerking, f.eks. en poly-hismerking i en Baculovirus-uttrykkingsvektor. Det poly-his-merkede DNA-et som koder for det innsatte antistoffet mot IgE/M1' kan deretter subklones inn i en SV40- drevet vektor som inneholder en seleksjonsmarkør som f.eks. DHFR for seleksjon av stabile kloner. Til slutt kan CHO-cellene transfekteres (som beskrevet ovenfor) med den SV40-drevne vektoren. Merkingen kan utføres som beskrevet ovenfor for å verifisere uttrykkingen. Kulturmediet som inneholdt det uttrykte poly-his-merkede antistoffet mot IgE/M1' kan så konsentreres og renses ved en hvilken som helst valgt fremgangsmåte, f.eks. ved Ni 2+ -kelataffinitetstkromatografi. 1 Antistoffet mot IgE/M1' kan også uttrykkes i CHO- og/eller COS-celler ved en forbigående uttrykkingsprosedyre eller i CHO-celler med en annen stabil uttrykkingsprosedyre. 20 Stabil uttrykking i CHO-celler utføres ved følgende prosedyre. Proteinene uttrykkes som en IgG-konstruksjon (immunadhesin), der kodingssekvensene for de løselige formene (f.eks. ekstracellulære domener) av de respektive proteinene fusjoneres til en sekvens for den konstante regionen av IgG1 som inneholder hengsel-, CH2- og CH3-domenet og/eller en poly-his-merket form Etter PCR-forsterking subklones de respektive DNA-ene i en CHO-uttrykkingsvektor ved standardmetoder som beskrevet i Ausubel et al., Current Protocols of Molecular Biology, Unit 3.16, John Wiley and Sons (1997). CHO-uttrykkingsvektorene konstrueres slik at de har kompatible restriksjonsseter i ' og 3' på DNA-et av interesse for å gjøre det enklere å transportere cdna. Vektoren som anvendes ved uttrykking i CHO-celler er som beskrevet i Lucas et al., Nucl. Acids Res. 24:9 ( (1996), og anvender SV40 tidlig promotor/forsterker til å drive uttrykkingen av det interessante cdna-et og dihydrofolatreduktase (DHFR). DHFR-utrykkingen gjør det mulig å velge for stabilt vedlikehold av plasmidet etter transfekteringen. 3 Tolv mikrogram av det ønskede plasmid-dna-et innføres i rundt millioner CHO-celler ved hjelp av kommersielt tilgjengelige trasnfeksjonsreagenser SUPERFECT (Quiagen), DOSPER eller FUGENE (Boehringer Mannheim).
164 162 Cellene dyrkes som beskrevet i Lucas et al., ovenfor. Rundt 3 x 7 celler blir frosset i en ampulle for ytterligere vekst og produksjon som beskrevet nedenfor Ampullene som inneholdt plasmid-dna tines ved å sette dem i vannbad og blandes på virvelmikser. Innholdet pipetteres i et sentrifugerør som inneholder ml medium og sentrifugeres ved 00 rpm i minutter. Supernatanten aspireres og cellene resuspenderes i ml selektivt medium (0,2 µm-filtrert PS20 med % 0,2 µm diafiltrert kalvefosterserum). Så porsjoneres cellene i en 0 ml sentrifugekolbe som inneholder 90 ml selektivt medium. Etter 1 2 dager overføres cellene til en 20 ml sentrifugekolbe fylt med ml selektivt vekstmedium og inkuberes ved 37 C. Etter ytterligere 2 til 3 dager blir 20 ml, 00 ml og 2000 ml sentrifugekolber utsådd med 3 x celler/ml. Cellemediet byttes ut med friskt medium ved sentrifugering og resuspensjon i produksjonsmediet. Et hvilket som helst egnet CHO-medium kan benyttes, men et produksjonsmedium som beskrevet i U.S. patent nr , utstedt 16. juni 1992 kan faktisk anvendes. En 3 l produksjonskolbe utsås med 1,2 x 6 celler/ml. På dag 0 bestemmes celleantall og ph. På dag 1 tas det prøve av kolben, og spyling med filtrert luft påbegynnes. På dag 2 tas det prøve av kolben, temperaturen forskyves til 33 C og det tas 30 ml 00 g/l glukose og 0,6 ml % antiskummiddel (f.eks. 3 % polydimetylsiloksanemulsjon, Dow Coming 36 Medical Grade Emulsion). I løpet av produksjonen justeres ph etter behov for å holde den rundt 7,2. Etter dager, eller inntil levedyktigheten falt under 70 %, høstes cellekulturen ved sentrifugering og filtrering gjennom et 0,22 µm filter. Filtratet ble enten lagret ved 4 C eller umiddelbart lastet på kolonner for rensing. For de poly-his-merkede konstruksjonene renses proteinene med en Ni-NTAkolonne (Qiagen). Før rensingen tilsettes det kondisjonerte mediet imidazol til en konsentrasjon på mm. Det kondisjonerte mediet pumpes på en 6 ml Ni- NTA-kolonne brakt i likevekt ved 4 C, i 20 mm Hepes, ph 7,4-buffer som inneholder 0,3 M NaCl og mm imidazol med strømningshastighet 4 ml/min. Etter påpumpingen vaskes kolonnen med mer likevektsbuffer og proteinet elueres med en likevektsbuffer som inneholder 0,2 M imidazol. Det høyrensede proteinet blir så avsaltet i en lagringsbuffer som inneholder mm Hepes, 0,14 M NaCl og 4 % mannitol, ph 6,8, med en 2 ml G2 Superfine (Pharmacia)-kolonne og lagret ved 80 C.
165 163 Immunadhesinkonstruksjoner (Fc-holdige) renses fra det kondisjonerte mediet som følger. Det kondisjonerte mediet pumpes på en ml protein-a-kolonne (Pharmacia) som er likevekstbehandlet i 20 mm Na-fosfatbuffer, ph 6,8. Etter lastingen vaskes kolonnen grundig med likevektsbufferen før den elueres med 0 mm sitronsyre, ph 3,. Det eluerte proteinet nøytraliseres umiddelbart ved å samle 1 ml fraksjoner i rør som inneholder 27 µl 1 M Tris-buffer, ph 9. Det høyrensede proteinet blir så avsaltet i lagringsbuffer som beskrevet ovenfor for de poly-his-merkede proteinene. Homogeniteten vurderes med SDS-polyakrylamidgeler og ved N- endeaminosyresekvensering ved Edman-nedbrytning. Deponering av materiale: De følgende materialene er deponert hos American Type Culture Collection, 801 University Blvd., Manassas, VA , USA (ATCC): Materiale ATCC-dep. nr. Deponeringsdato 7A6.18 PTA mars C PTA mars G4.6.2 PTA mars H4.12. PTA mars H4.6.9 PTA mars A PTA mars A11.6. PTA mars D PTA mars C PTA mars B PTA mars E PTA mars Disse deponeringene ble gjort ifølge bestemmelsene i Budapest-konvensjonen om internasjonal anerkjennelse av deponering av mikroorganismer i forbindelse med
166 164 patentprosedyrer og forskriftene i henhold til den (Budapest-konvensjonen). Dette sikrer at det opprettholdes en levedyktig kultur for deponeringene i tretti (30) år fra deponeringsdatoen, og minst fem () år etter den siste forespørselen om en prøve. Deponeringene vil bli gjort tilgjengelig av ATCC i henhold til vilkårene i Budapest-konvensjonen, og forutsatt en avtale mellom Genentech, Inc. og ATCC, som sikrer publikum permanent og ubegrenset tilgjengelighet for avkommet av kulturen i deponeringen enten etter at det relevante amerikanske patentet er utstedt eller etter at eventuelle amerikanske eller utenlandske patentsøknader er offentliggjort hvis dette skjer før, og sikrer at avkommet er tilgjengelig for dem som har rett til dette etter bedømmelsen til U.S. Commissioner of Patents and Trademarks ifølge 3 USC 122 og kommissærens regler ifølge dette (inkludert 37 CFR 1.14 med særlig henvisning til 886 OG 638) Oppdragstakeren i den foreliggende søknaden har sagt seg enig i at hvis en kultur av materialene i deponeringen skulle dø eller bli borte eller ødelagt under dyrking ved egnede forhold, blir materialene erstattet umiddelbart etter at det er mottatt varsel med en annen av kultur av samme materiale. Tilgjengeligheten til det deponerte materialet skal ikke tolkes som en tillatelse til å praktisere oppfinnelsen i strid med rettighetene som er tildelt med hjemmel som noen som helst regjering har gitt i samsvar med patentlovene sine. 2 Den foregående skriftlige beskrivelsen anses å være tilstrekkelig for å gjøre en fagmann på området i stand til å praktisere oppfinnelsen. Den foreliggende oppfinnelsen skal ikke begrenses i omfang av eksemplet presentert i dette dokumentet. Faktisk vil forskjellige modifikasjoner av oppfinnelsen i tillegg til de som er vist og beskrevet i dette dokumentet bli åpenbare for fagfolk ut fra den foregående beskrivelsen.
167 16 PATENTKRAV 1. Antistoff mot IgE/M1' som spesifikt binder en epitop i M1'-segmentet av IgE definert ved aminosyrerest 317 til 31 i SEQ ID NO:1 og som utløser apoptose i IgE-uttrykkende B-celler. 2. Antistoffet ifølge krav 1, som kan binde IgE som stammer fra menneske, rhesusape eller krabbemakak. 3. Antistoffet ifølge krav 1 eller krav 2, som spesifikt desimerer IgEproduserende B-celler når en terapeutisk effektiv mengde administreres til et pattedyr in vivo, og eventuelt senker det totale serum-ige eller senker fritt serum-ige; der IgE eventuelt er allergenspesifikk Antistoffet ifølge krav 3, som er kimært, humanisert eller humant Antistoffet ifølge et hvilket som helst av de foregående kravene som spesifikt binder den samme epitopen som bindes av et antistoff valgt fra gruppen som består av: 47H4 (ATTC dep. nr. PTA-8270), 7A6 (ATTC dep. nr. PTA-8268), 26A11 (ATTC dep. nr. PTA-8262), 47H4v som omfatter en variabel lett kjede med aminosyresekvensen som er vist som SEQ ID NO:31 (fig. 6C), og en variabel tung kjede med aminosyresekvensen som er vist som SEQ ID NO:41 (fig. 6F), 7A6v1 som omfatter en variabel lett kjede med aminosyresekvensen som er vist som SEQ ID NO:28 (fig. 6B) og en variabel tung kjede med aminosyresekvensen som er vist som SEQ ID NO:37 (fig. 6E), og 26A11 v6 som omfatter en variabel lett kjede med aminosyresekvensen som er vist som SEQ ID NO:24 (fig. 6A), og en variabel tung kjede med aminosyresekvensen som er vist som SEQ ID NO:3 (fig. 6D) Antistoffet ifølge krav der epitopen tilsvarer et peptid valgt fra gruppen som består av: peptid 4 (SEQ ID NO:8), peptid (SEQ ID NO:9), peptid 7 (SEQ ID NO:11) eller peptid 8 (SEQ ID NO:12) og der epitopen fortrinnsvis tilsvarer peptid 4 (SEQ ID NO:8) Antistoffet ifølge et hvilket som helst av de foregående kravene som binder med en Scatchard-bindingsaffinitet som tilsvarer bindingsaffiniteten til det murine antistoffet 47H4 mot IgE/M1' (ATTC-dep. nr. PTA-8270) og eventuelt med en affinitet på mellom 0,30 og 0,83 nm.
168 Antistoffet ifølge et hvilket som helst av de foregående kravene som spesifikt binder et M1'-segment av IgE med en Scatchard-bindingsaffinitet som tilsvarer bindingsaffiniteten til humanisert antistoff 47H4v mot IgE/M1' som omfatter en variabel lett kjede med aminosyresekvensen som er vist som SEQ ID NO:31 (fig. 6C), og en variabel tung kjede med aminosyresekvensen som er vist som SEQ ID NO:41 (figur 6F), og eventuelt med en affinitet på rundt 1, nm Antistoffet ifølge et hvilket som helst av de foregående kravene som omfatter HVR fra den tunge og lette kjeden til et antistoff eller antigenbindende fragment av dette valgt fra gruppen som består av: 26A11 som vist i SEQ ID NO: 21 (fig. 6A) og SEQ ID NO: 33 (fig. 6D), 26A11v.1-16 som vist i SEQ ID NO: (fig. 6A) og SEQ ID NO: 34 og 3 (fig. 6D), 7A6 som vist i SEQ ID NO: 27 (fig. 6B) og SEQ ID NO: 36 (fig. 6E), 7A6v1 som vist i SEQ ID NO: 28 (fig. 6B) og SEQ ID NO: 37 (fig. 6E), 47H4 som vist i SEQ ID NO: 29 (fig. 6C) og SEQ ID NO: 38 (fig. 6F) og 47H4v1-6 som vist i SEQ ID NO: 30 og 31 (fig. 6C) og SEQ ID NO: (fig. 6F), og som fortrinnsvis omfatter HVR fra den tunge og lette kjeden eller et antistoff eller et antigenbindende fragment av dette blant 47H4v1 6 som vist i SEQ ID NO: 30 og 31 (fig. 6C) og SEQ ID NO: (fig. 6F).. Antistoffet ifølge krav 9 som omfatter de variable regionene av den tunge og den lette kjeden til antistoffet eller et antigenbindende fragment av dette valgt fra gruppen som består av: 26A11 som vist i SEQ ID NO: 21 (fig. 6A) og SEQ ID NO: 33 (fig. 6D), 26A11v.1-16 som vist i SEQ ID NO: (fig. 6A) og SEQ ID NO: 34 og 3 (fig. 6D), 7A6 som vist i SEQ ID NO: 27 (fig. 68) og SEQ ID NO: 36 (fig. 6E), 7A6v1 som vist i SEQ ID NO: 28 (fig. 6B) og SEQ ID NO: 37 (fig. 6E), 47H4 som vist i SEQ ID NO: 29 (fig. 6C) og SEQ ID NO: 38 (fig. 6F) og 47H4v1-6 som vist i SEQ ID NO: 30 og 31 (fig. 6C) og SEQ ID NO: (fig. 6F) Antistoffet ifølge krav, som er afukosylert. 12. Peptid valgt fra gruppen som består av: peptid 4 (SEQ ID NO:8), peptid (SEQ ID NO:9), peptid 7 (SEQ ID NO:11) og peptid 8 (SEQ ID NO:12) Sammensetning som omfatter antistoffet ifølge et hvilket som helst av kravene 1 11 i kombinasjon med minst én farmasøytisk aksepterbar bærer.
169 Sammensetningen ifølge krav 13 som også omfatter ett eller flere legemidler valgt fra gruppen som består av: antistoff mot IgE, antihistamin, bronkodilator, glukokortikoid, NSAID, TNF-antagonist, integrinantagonist, immunundertrykkende middel, IL4-antagonist, IL13-antagonist, dobbel IL4-/IL13-antagonist, DMARD, antistoff som binder til en B-celleoverflatemarkør, og BAFF-antagonist Isolert nukleinsyre som koder for et antistoff eller antigenbindende fragment av dette som omfatter HVR fra den tunge og den lette kjeden til et apoptotisk antistoff mot IgE/M1' eller et antigenbindende fragment av dette valgt fra gruppen som består av: 26A11 som vist i SEQ ID NO: 21 (fig. 6A) og SEQ ID NO: 33 (fig. 6D), 26A11 v1-16 som vist i SEQ ID NO: (fig. 6A) og SEQ ID NO: 34 og 3 (fig. 6D), 7A6 som vist i SEQ ID NO: 27 (fig. 6B) og SEQ ID NO: 36 (fig. 6E), 7A6v1 som vist i SEQ ID NO: 28 (fig. 6B) og SEQ ID NO: 37 (fig. 6E), 47H4 som vist i SEQ ID NO: 29 (fig. 6C) og SEQ ID NO: 38 (fig. 6F) og 47H4v1-6 som vist i SEQ ID NO: 30 og 31 (fig. 6C) og SEQ ID NO: (fig. 6F) Antistoffet ifølge krav 1 som også omfatter nukleinsyre som koder for de variable regionene av den tunge og den lette kjeden til antistoffsekvensen valgt fra gruppen som består av: 26A11 som vist i SEQ ID NO: 21 (fig. 6A) og SEQ ID NO: 33 (fig. 6D), 26A11v1-16 som vist i SEQ ID NO: (fig. 6A) og SEQ ID NO: 34 og 3 (fig. 6D), 7A6 som vist i SEQ ID NO: 27 (fig. 6B) og SEQ ID NO: 36 (fig. 6E), 7A6v1 som vist i SEQ ID NO: 28 (fig. 6B) og SEQ ID NO: 37 (fig. 6E), 47H4 som vist i SEQ ID NO: 29 (fig. 6C) og SEQ ID NO: 38 (fig. 6F) og 47H4v1-6 som vist i SEQ ID NO: 30 og 31 (fig. 6C) og SEQ ID NO: (fig. 6F). 17. Nukleinsyren ifølge krav 16, der antistoffet det kodes for er afukosylert Vektor der nukleinsyren ifølge et hvilket som helst av kravene 1 til 17 er operativt koblet. 19. Vertscelle som omfatter en vektor ifølge krav Vertscellen ifølge krav 19 som er et en pattedyrcelle og som fortrinnsvis er en eggstokkcelle fra kinesisk dverghamster. 21. Prosess for å fremstille et apoptotisk antistoff mot IgE/M1' eller funksjonelt fragment, som omfatter å dyrke vertscellen ifølge krav 19 eller
170 168 krav 20 under forhold som er egnet for uttrykking av antistoffet eller fragmentet og utvinning av antistoffet eller fragmentet. 22. Produksjonsgjenstand som omslutter sammensetningen ifølge krav 13 eller 14 og et pakningsvedlegg som anviser anvendelsen for behandling av en IgE-formidlet forstyrrelse. 23. Gjenstanden ifølge krav 22 som er (i) et glass eller (ii) en forhåndsfylt sprøyte, som eventuelt også omfatter en injeksjonsanordning, f.eks. en autoinjektor Antistoff ifølge et hvilket som helst av kravene 1 til 11 for anvendelse i en fremgangsmåte for å desimere IgE-produserende B-celler spesifikt, idet fremgangsmåten omfatter å administrere en terapeutisk effektiv mengde av antistoff mot IgE/M1 til et pattedyr og antistoffet eventuelt har ADCC-aktivitet Antistoff ifølge et hvilket som helst av kravene 1 til 11 for anvendelse i en fremgangsmåte for å behandle en IgE-formidlet forstyrrelse, der fremgangsmåten omfatter å administrere en terapeutisk effektiv mengde av antistoffet mot IgE/M1'. 26. Antistoffet for anvendelse i en fremgangsmåte for behandling ifølge krav 2, der: (i) den IgE-formidlede forstyrrelsen er valgt fra gruppen som består av: allergisk rhinitt, allergisk astma, ikke-allergisk astma, atopisk dermatitt, allergisk gastroenteropati, anafylaksi, elveblest, matallergier, allergisk bronkopulmonal aspergillose, parasittsykdommer, interstitiell cystitt, hyper-ige-syndrom, ataksitelangiektasi, Wiskott-Aldrich-syndrom, lymphoplasia athymica, IgE-myelom, transplantat-mot-vert-reaksjon og allergisk purpura og/eller (ii) antistoffet skal administreres i kombinasjon med en terapeutisk effektiv mengde av minst ett legemiddel valgt fra gruppen som består av: antistoff mot IgE, antihistamin, bronkodilator, glukokortikoid, NSAID, slimhinneavsvellende legemiddel, hostedempende legemiddel, analgetikum, TNF-antagonist, integrinantagonist, immunundertrykkende middel, IL4-antagonist, IL13- antagonist, dobbel IL4/IL13-antagonist, DMARD, antistoff som binder til en B- celleoverflatemarkør og BAFF-antagonist. (iii) antistoffet skal administreres i et kombinert behandlingsregime som omfatter å administrere en terapeutisk effektiv mengde av antistoff mot
171 169 IgE/M1' før, samtidig med eller etter administreringen av en kjent fremgangsmåte for å behandle allergiske forstyrrelser. 27. Antistoffet for anvendelse i en fremgangsmåte for behandling ifølge krav 26(iii), der: (a) den kjente behandlingen for allergisk forstyrrelse omfatter å administrere et antihistamin for antistoff mot IgE, en bronkodilator, et glukokortikoid, et ikkesteroid antiinflammatorisk legemiddel, et immunundertrykkende middel, en IL4- antagonist, en IL13-antagonist, en dobbel IL4/IL13-antagonist, en slimhinneavsvellende middel, et hostedempende legemiddel eller et analgetikum; eller (b) den kjente behandlingen for allergisk forstyrrelse omfatter et behandlingsregime for allergendesensibilisering Antistoffet for anvendelse i en fremgangsmåte for behandling ifølge et hvilket som helst av kravene 24 til 27, der fremgangsmåten er for å hindre eller redusere allergenutløst IgE-produksjon Anvendelse av et antistoff ifølge et hvilket som helst av kravene 1 11 i produksjon av et legemiddel for behandling av en IgE-formidlet forstyrrelse Anvendelsen ifølge krav 29, der: (i) den IgE-formidlede forstyrrelsen er valgt fra gruppen som består av: allergisk rhinitt, allergisk astma, ikke-allergisk astma, atopisk dermatitt, allergisk gastroenteropati, anafylaksi, elveblest, matallergier, allergisk bronkopulmonal aspergillose, parasittsykdommer, interstitiell cystitt, hyper-ige-syndrom, ataksitelangiektasi, Wiskott-Aldrich-syndrom, lymphoplasia athymica, IgE-myelom, transplantat-mot-vert-reaksjon og allergisk purpura; og/eller (ii) antistoffet skal administreres i kombinasjon med en terapeutisk effektiv mengde av minst ett legemiddel valgt fra gruppen som består av: antistoff mot IgE, antihistamin, bronkodilator, glukokortikoid, NSAID, slimhinneavsvellende legemiddel, hostedempende legemiddel, analgetikum, TNF-antagonist, integrinantagonist, immunundertrykkende middel, IL4-antagonist, IL13- antagonist, dobbel IL4/IL13-antagonist, DMARD, antistoff som binder til en B- celleoverflatemarkør og BAFF-antagonist.
172 Murint hybridom deponert hos ATCC den 21. mars 2007 valgt fra gruppen som består av: PTA-8260, PTA-8261, PTA-8262, PTA-8263, PTA-8264, PTA-826, PTA-8266, PTA-8267, PTA-8268, PTA-8269, PTA Antistoff som skilles ut av hybridomet ifølge krav Transgent dyr som uttrykker det humane M1'-segmentet av IgE som omfatter rest 317 til 31 i SEQ ID NO:1.
173 1 Sekvensliste <1> Genent ech, Inc. Wu, Lawren Balazs, Mer cedesz Bright bill, Hans Chan, Andr ew C. Chen, Yvonne Man-yee Chunt har apai, Anan Dennis, Mark Wong, Ter ence <120> Apoptotic Anti-IgE Antibodies <130> P2474R1 WO <141> <> US 60/ 896, 339 <11> <160> 112 <2> 1 <211> 431 <212> PRT <213> Homo sapiens <220> <221> Ot her... <223> human IgE <400> 1
174 2 NO/EP
175 3 <2> 2 <211> 432 <212> PRT <213> Macaca mulatta <220> <221> Other... <223> rhesus IgE <400> 2
176 4 NO/EP
177 <2> 3 <211> 431 <212> PRT <213> Macaca fascicularis <220> <221> Other... <223> Cyno IgE <400> 3
178 6 <2> 4 <211> 4 <212> PRT <213> Homo sapiens <220> <221> Other... <223> Human IgE fragment <400> 4
179 7 <2> <211> 1 <212> PRT <213> Artificial sequence <220> <223> M1' pepide 1 <400> <2> 6 <211> 1 <212> PRT <213> Artificial sequence <220> <223> M1' peptide 2 <400> 6 <2> 7 <211> 1 <212> PRT <213> Artificial sequence <220> <223> M1' peptide 3 <400> 7 <2> 8 <211> 1 <212> PRT <213> Artificial sequence <220> <223> M1' pept i de 4 <400> 8 <2> 9 <211> 1 <212> PRT <213> Artificial sequence <220>
180 8 <223> M1' peptide <400> 9 <2> <211> 1 <212> PRT <213> Artificial sequence <220> <223> M1 peptide 6 <400> <2> 11 <211> 1 <212> PRT <213> Artificial sequence <220> <223> M1' pept i de 7 <400> 11 <2> 12 <211> 14 <212> PRT <213> Artificial sequence <220> <223> M1' peptide 8 <400> 12 <2> 13 <211> 1 <212> PRT <213> Artificial sequence <220> <223> M1' peptide 9 <400> 13 <2> 14 <211> 1
181 9 <212> PRT <213> Artificial sequence <220> <223> M1' peptide <400> 14 <2> 1 <211> 1 <212> PRT <213> Artificial sequence <220> <223> M1' peptide 11 <400> 1 <2> 16 <211> 1 <212> PRT <213> Artificial sequence <220> <223> M1' peptide 12 <400> 16 <2> 17 <211> 1 <212> PRT <213> Artificial sequence <220> <223> M1' peptide 13 <400> 17 <2> 18 <211> 1 <212> PRT <213> Artificial sequence <220> <223> M1' peptide 14 <400> 18
182 <2> 19 <211> 1 <212> PRT <213> Artificial sequence <220> <223> M1' peptide 1 <400> 19 <2> 20 <211> 8 <212> PRT <213> Homo sapi ens <220> <221> Other... <223> Human kappa I antibody light chain <400> 20 <2> 21 <211> 8 <212> PRT <213> Mus musculus <220> <221> Other... <223> Marine 26A11 antibody light chain <400> 21
183 11 <2> 22 <211> 8 <212> PRT <213> Artificial sequence <220> <223> Humanized 26A11 v 1, 4 antibody light chai n <400> 22 <2> 23 <211> 8 <212> PRT <213> Artificial sequence <220> <223> humanized 26A11 v2, ant i body light chai n <400> 23
184 12 <2> 24 <211> 8 <212> PRT <213> Artificial sequence <220> <223> humanized 26A11 V3, 6 antibody light chai n <400> 24 <2> 2 <211> 8 <212> PRT <213> Artificial sequence <220> <223> Humanized 26A11 V13, 1 antibody light chain <400> 2
185 13 <2> 26 <211> 8 <212> PRT <213> Artificial sequence <220> <223> Humanized 26A11 V14, 16 antibody light chai n <400> 26 <2> 27 <211> 114 <212> PRT <213> Mus msculus <220> <221> Other... <223> Murine 7A6 antibody light chain <400> 27
186 14 <2> 28 <211> 114 <212> PRT <213> Artificial sequence <220> <223> Humanized 7A6 V. 1 antibody light chain <400> 28 <2> 29 <211> 113 <212> PRT <213> Mus musculus <220> <221> Other... <223> Humanized 47H4 antibody light chain <400> 29
187 1 <2> 30 <211> 113 <212> PRT <213> Artificial sequence <220> <223> Humanized 47H4 V. 1, 3 antibody light chain <400> 30 <2> 31 <211> 113 <212> PRT <213> Artificial sequence <220> <223> Humanized 47H4 V ant i body light chain <400> 31
188 16 <2> 32 <211> 113 <212> PRT <213> Homo sapi ens <220> <221> Other... <223> Human III antibody heavy chain <400> 32 <2> 33 <211> 112 <212> PRT <213> Mus musculus <220> <221> Other...
189 17 <223> Murine 26A11 antibody heavy chain <400> 33 <2> 34 <211> 112 <212> PRT <213> Artificial sequence <220> <223> Humanized 26A11 V. 1-3, 13, 14 antibody heavy chain <400> 34 <2> 3 <211> 112 <212> PRT <213> Artificial sequence <220> <223> Humanized 26A11 V. 4-6, 1, 16 antibody heavy chain <400> 3
190 18 <2> 36 <211> 122 <212> PRT <213> Mus musculus <220> <221> Other... <223> Murine 7A6 anti body heavy chain <400> 36 <2> 37 <211> 122 <212> PRT <213> Artificial sequence <220> <223> Humanized 7A6 V. antibody heavy chain
191 19 <400> 37 <2> 38 <211> 117 <212> PRT <213> Mus musculus <220> <221> Other... <223> Murine 47H4 antibody heavy chain <400> 38 <2> 39 <211> 117 <212> PRT <213> Artificial sequence
192 20 <220> <223> Humanized 47H4 V. 1,2 antibody heavy chai n <400> 39 <2> 40 <211> 117 <212> PRT <213> Artificial sequence <220> <223> Humanized 47H4 V.3,4 antibody heavy chai n <400> 40 <2> 41 <211> 117 <212> PRT <213> Artificial sequence
193 21 <220> <223> Humanized 47H4 V. antibody heavy chain <400> 41 <2> 42 <211> 117 <212> PRT <213> Artificial sequence <220> <223> Humanized 47H4 V.6 antibody heavy chain <400> 42 <211> 446 <212> PRT <213> Mus musculus <220> <221> Other...
194 22 <223> Murine 7A6 antibody heavy chain full length <400> 43
195 23 <2> 44 <211> 220 <212> PRT <213> Mus musculus <220> <221> Other... <223> Murine 7A6 antibody light chain full length <400> 44
196 24 <2> 4 <211> 442 <212> PRT <213> Mus musculus <220> <221> Other... <223> Murine 47H4 antibody heavy chain full length <400> 4
197 2 <2> 46 <211> 219 <212> PRT <213> Mus musculus <220> <221> Other... <223> Murine 47H4 light chain full length <400> 46
198 26 <2> 47 <211> 436 <212> PRT <213> Mus musculus <220> <221> Other... <223> Murine 26A11 antibody heavy chain full lengt'h <400> 47
199 27 <2> 48 <211> 214 <212> PRT <213> Mus musculus <220> <221> Other...
200 28 <223> Murine 26A11 antibody light chain full length <400> 48 <2> 49 <211> 449 <212> PRT <213> Mus musculus <220> <221> Other... <223> Murine 4C1 antibody heavy chain full length <400> 49
201 29 NO/EP
202 30 <2> 0 <211> 219 <212> PRT <213> Mus musculus <220> <221> Other... <223> Murine 4C1 antibody light chain full length <400> 0
203 31 <2> 1 <211> 446 <212> PRT <213> Mus musculus <220> <221> Other... <223> Murine 28E9 antibody heavy chain full length <400> 1
204 32 <2> 2 <211> 214 <212> PRT <213> Mus musculus <220> <221> Other... <223> Murine 28E9 antibody light chain full length <400> 2
205 33 <2> 3 <211> 442 <212> PRT <213> Mus musculus <220> <221> Other... <223> Murine 1C11 antibody heavy chain full length <400> 3
206 34 <2> 4 <211> 220 <212> PRT <213> Mus musculus <220> <221> Other... <223> Murine 1C11 antibody light chain full length <400> 4
207 3 <2> <211> 2 <212> PRT <213> Homo sapiens <220> <221> Other... <223> VH Human consesus framework - H1 <400> <2> 6 <211> 14 <212> PRT <213> Homo sapiens <220> <221> Other... <223> VH Human consensus framework - H2
208 36 <400> 6 <2> 7 <211> 32 <212> PRT <213> Homo sapiens <220> <221> Other... <223> VH Human consensus framework - H3 <400> 7 <2> 8 <211> 11 <212> PRT <213> Homo sapiens <220> <221> Other... <223> VH Human consensus framework - H4 <400> 8 <2> 9 <211> 23 <212> PRT <213> Homo sapiens <220> <221> Other... <223> VL Human consensus framework - L1 <400> 9 <2> 60 <211> 1 <212> PRT <213> Homo sapiens
209 37 <220> <221> Other... <223> VL Human consensus framework - L2 <400> 60 <2> 61 <211> 32 <212> PRT <213> Homo sapiens <220> <221> Other... <223> VL Human consensus framework - L3 <400> 61 <2> 62 <211> 11 <212> PRT <213> Homo sapiens <220> <221> Other... <223> VL Human consensus framework - L4 <400> 62 <2> 63 <211> 26 <212> DNA <213> Artificial sequence <220> <223> Forward cloning primer used in example 1 <400> 63 ccgcccaccg tgaagatcttacagtc 26 <2> 64 <211> 3
210 38 <212> DNA <213> Artificial sequence <220> <223> Reverse cloning primer used in example 1 <400> 64 cggct cgaga ct aggcgt gg ggctggagga cgt t g 3 <2> 6 <211> 42 <212> PRT <213> Artificial sequence <220> <223> IgE Ch3-Ch4-M-Fc fusion <400> 6
211 39 NO/EP
212 40 <2> 66 <211> 2 <212> PRT <213> Mus musculus <220> <221> Other... <223> N-terminal sequence, heavy chain 7AG <400> 66 <2> 67 <211> 2 <212> PRT <213> Mus musculus <220> <221> Other... <223> N-terminal sequence, light chain 7AG <400> 67
213 41 <2> 68 <211> 2 <212> PRT <213> Mus musculus <220> <221> Other... <223> N-terminal sequence, heavy chain 1C11 <400> 68 <2> 69 <211> 2 <212> PRT <213> Mus musculus <220> <221> Other... <223> N-terminal sequence, light chain 1C11 <400> 69 <2> 70 <211> 2 <212> PRT <213> Mus musculus <220> <221> Other... <223> N-terminal sequence, heavy chai n 47H4 <400> 70 <2> 71 <211> 21 <212> PRT
214 42 <213> Mus musculus <220> <221> Other... <223> N-terminal sequence, light chain 47H4 <400> 71 <2> 72 <211> 2 <212> PRT <213> Mus musculus <220> <221> Other... <223> N-terminal sequence, heavy chain 26A11 <400> 72 <2> 73 <211> 2 <212> PRT <213> Mus musculus <220> <221> Other... <223> N-terminal sequence, light chain 26A11 <400> 73 <2> 74 <211> 19 <212> PRT <213> Mus musculus <220> <221> Other... <223> N-terminal sequence, heavy chai n 4C1 <400> 74
215 43 <2> 7 <211> 24 <212> PRT <213> Mus musculus <220> <221> Other... <223> N-terminal sequence, light chain 4C1 <400> 7 <2> 76 <211> 2 <212> PRT <213> Mus musculus <220> <221> Other... <223> N-terminal sequence, heavy chain 28E9 <400> 76 <2> 77 <211> 24 <212> PRT <213> Mus musculus <220> <221> Other... <223> N-terminal sequence, light chain 28E9 <400> 77 <2> 78 <211> 46 <212> DNA
216 44 <213> Artificial sequence <220> <223> FOR. Bsi W 7AG HC f or war d primer <400> 78 tcgacgtacg ct caggt t ca gct gcagcaa t ct ggggct g agctgg 46 <2> 79 <211> 39 <212> DNA <213> Artificial sequence <220> <223> FOR. EcoRV 7A6 LC forward primer <400> 79 gatcgatat cgtgatgtccc agt ct ccct c ct ccct aac 39 <2> 80 <211> 46 <212> DNA <213> Artificial sequence <220> <223> FOR. Bsi W 1C11 HC forward primer <400> 80 t cgacgt acg ct caggt t ca at t gcagcag t ct ggggct g agctgg 46 <2> 81 <211> 39 <212> DNA <213> Artificial sequence <220> <223> FOR. EcoRV 1C11 LC forward primer <400> 81 gat cgat at c gtaatgtctc agtctccttc ctccctagc 39 <2> 82 <211> 46 <212> DNA <213> Artificial sequence <220> <223> 9. 1 HCF. Bsi W 47H4 HC forward primer <400> 82 t cgacgt acg ctgaggtgaa gt t ggt ggag t ct gggggag gcttag 46
217 4 <2> 83 <211> 39 <212> DNA <213> Artificial sequence <220> <223> 47H4LCF. EcoRV 47H4 LC forward primer <400> 83 gat cgat at c gt gct gact c agactccact ctccctgcc 39 <2> 84 <211> 43 <212> DNA <213> Artificial sequence <220> <223> C7F7HCF. Bsi W 26A11 HC forward primer <400> 84 t cgacgt acg ct gaggt cca gct ccagcag t ct ggacct g agc 43 <2> 8 <211> 37 <212> DNA <213> Artificial sequence <220> <223> 2B4LCF. EcoRV 26A11 LC forward primer <400> 8 gat cgat at c cagat gaccc aaact acat c ct ccct g 37 <2> 86 <211> 4 <212> DNA <213> Artificial sequence <220> <223> 2G6HCF. Bsi W 4C1 HC forward primer <400> 86 t cgacgt acg ct cagat cca gttggtgcag t ct ggacct g agct g 4 <2> 87 <211> 41 <212> DNA <213> Artificial sequence <220>
218 46 <223> 9CLCF. EcoRV 4C1 LC forward primer <400> 87 gatcgatatc gtgatgacgc agact ccact cactttgtcgg 41 <2> 88 <211> 46 <212> DNA <213> Artificial sequence <220> <223> 9.1HCF BsiW 28E9 HC forward primer <400> 88 t cgacgt acg ct gaggt gaa gct ggt ggag t ct gaaggag gct t gg 46 <2> 89 <211> 39 <212> DNA <213> Artificial sequence <220> <223> E. LCF. EcoRV 28E9 LC forward primer <400> 89 gat cgat at cagatgaccc agt ct ccagc ct ccct at c 39 <2> 90 <211> 4 <212> DNA <213> Artificial sequence <220> <223> Heavy Chai n Primer (HCR) <400> 90 ct ggacaggg atccagagtt t ccaggt cact gt cact ggct caggg 4 <2> 91 <211> 39 <212> DNA <213> Artificial sequence <220> <223> Light Chain Primer (LCR) <400> 91 ct gt aggt gc t gt ct t t gct gt cct gat ca gtccaactg 39 <2> 92 <211> 38
219 47 <212> DNA <213> Artificial sequence <220> <223> REV. Apal 7A6 HC reverse primer <400> 92 accgatgggc ccttggtgga ggctgaagag act gt gag 38 <2> 93 <211> 43 <212> DNA <213> Artificial sequence <220> <223> REV. Kpnl 7A6 LC reverse primer <400> 93 ccttggtacc ccct ccgaac gtgtacggat agctataata ttgg 43 <2> 94 <211> 37 <212> DNA <213> Artificial sequence <220> <223> REV. Apal 1C11 HC reverse primer <400> 94 gaccgatggg cccttggtgg aggctgagga gactgtg 37 <2> 9 <211> 38 <212> DNA <213> Artificial sequence <220> <223> REV. Kpnl 1C11 LC reverse primer <400> 9 ccttggtacc ccct ccgaac gt gt acggat agct at aa 38 <2> 96 <211> 37 <212> DNA <213> Artificial sequence <220> <223> 47H4HCR. Apal 47H4 HC reverse primer <400> 96
220 48 t gggccct t g gtggaggctgg aggagacggt gactgag 37 <2> 97 <211> 21 <212> DNA <213> Artificial sequence <220> <223> 47H4LCR. Kpnl 47H4 LC reverse primer <400> 97 ttccaacttg gtacctccac c 21 <2> 98 <211> 44 <212> DNA <213> Artificial sequence <220> <223> 7G8HCR. Apal 26A11 HC reverse primer <400> 98 gaccgatggg cccttggtgg argctgcaga gacagtgacc agag 44 <2> 99 <211> 21 <212> DNA <213> Artificial sequence <220> <223> 47H4LCR. Kpnl 26A11 LC reverse primer <400> 99 ttccaacttg gt acct ccac c 21 <2> 0 <211> 40 <212> DNA <213> Artificial sequence <220> <223> 4C1.HCR. Apal 4C1 HC reverse primer <400> 0 cgatgggccc ttggtggarg ckgaggagac ggtgagaatg 40 <2> 1 <211> 18 <212> DNA <213> Artificial sequence
221 49 <220> <223> C2LCR. Kpnl 4C1 LC reverse primer <400> 1 agcttggtac cccctccg 18 <2> 2 <211> 40 <212> DNA <213> Artificial sequence <220> <223> 28E9. HC. REV. Apal 28E9 HC reverse primer <400> 2 cgatgggccc ttggtggagg ctgaggagac ggcgactgag 40 <2> 3 <211> 22 <212> DNA <213> Artificial sequence <220> <223> 1D. 2LCR, Kpnl 28E9 LC r ever se primer <400> 3 ttccaacttg gtacccgagc cg 22 <2> 4 <211> 33 <212> DNA <213> Mus musculus <400> 4 cctatttaca ttggtatctg cagaagccag gcc 33 <2> <211> 33 <212> DNA <213> Artificial sequence <220> <223> Reverse mutagenesis primer <400> ggcctggctt ctgcagatac caat gt aaat agg 33 <2> 6 <211> 22 <212> DNA
222 0 <213> Artificial sequence <220> <223> M1' screening WT forward primer <400> 6 ggccaaagac cctaagacag t c 22 <2> 7 <211> 20 <212> DNA <213> Artificial sequence <220> <223> M1' scr eeni ng hum1' forward primer <400> 7 gggctggctg gcggctccgc 20 <2> 8 <211> 22 <212> DNA <213> Artificial sequence <220> <223> M1' screening WT reverse primer <400> 8 ctatgccctg gtctggaaga tg 22 <2> 9 <211> 2 <212> DNA <213> Artificial sequence <220> <223> Left ar m f or war d primer <400> 9 tgtctggtgg tggacctgga aagcg 2 <2> 1 <211> 2 <212> DNA <213> Artificial sequence <220> <223> Left arm reverse primer <400> 1 t cct cgct ct cct cct ct gg tggtg g 2
223 1 <2> 111 <211> 2 <212> DNA <213> Artificial sequence <220> <223> Right arm for ward primer <400> 111 ccat gcaacc t agt at cct a ttctc c 2 <2> 112 <211> 2 <212> DNA <213> Artificial sequence <220> <223> Right ar m reverse primer <400> 112 ctttatacag gagaacctag cccag 2
224 1 NO/EP
225 2 NO/EP
226 3 NO/EP
227 4 NO/EP
228 NO/EP
229 6 NO/EP
230 7 NO/EP
231 8 NO/EP
232 9 NO/EP
233 NO/EP
234 11 NO/EP
235 12 NO/EP
236 13 NO/EP
237 14 NO/EP
238 1 NO/EP
239 16 NO/EP
240 17 NO/EP
241 18 NO/EP
242 19 NO/EP
243 20 NO/EP
244 21 NO/EP
245 22 NO/EP
246 23 NO/EP
247 24 NO/EP
248 2 NO/EP
249 26 NO/EP
250 27 NO/EP
251 28 NO/EP
252 29 NO/EP
253 30 NO/EP
254 31 NO/EP
255 32 NO/EP
256 33 NO/EP
257 34 NO/EP
258 3 NO/EP
259 36 NO/EP
260 37 NO/EP
261 38 NO/EP
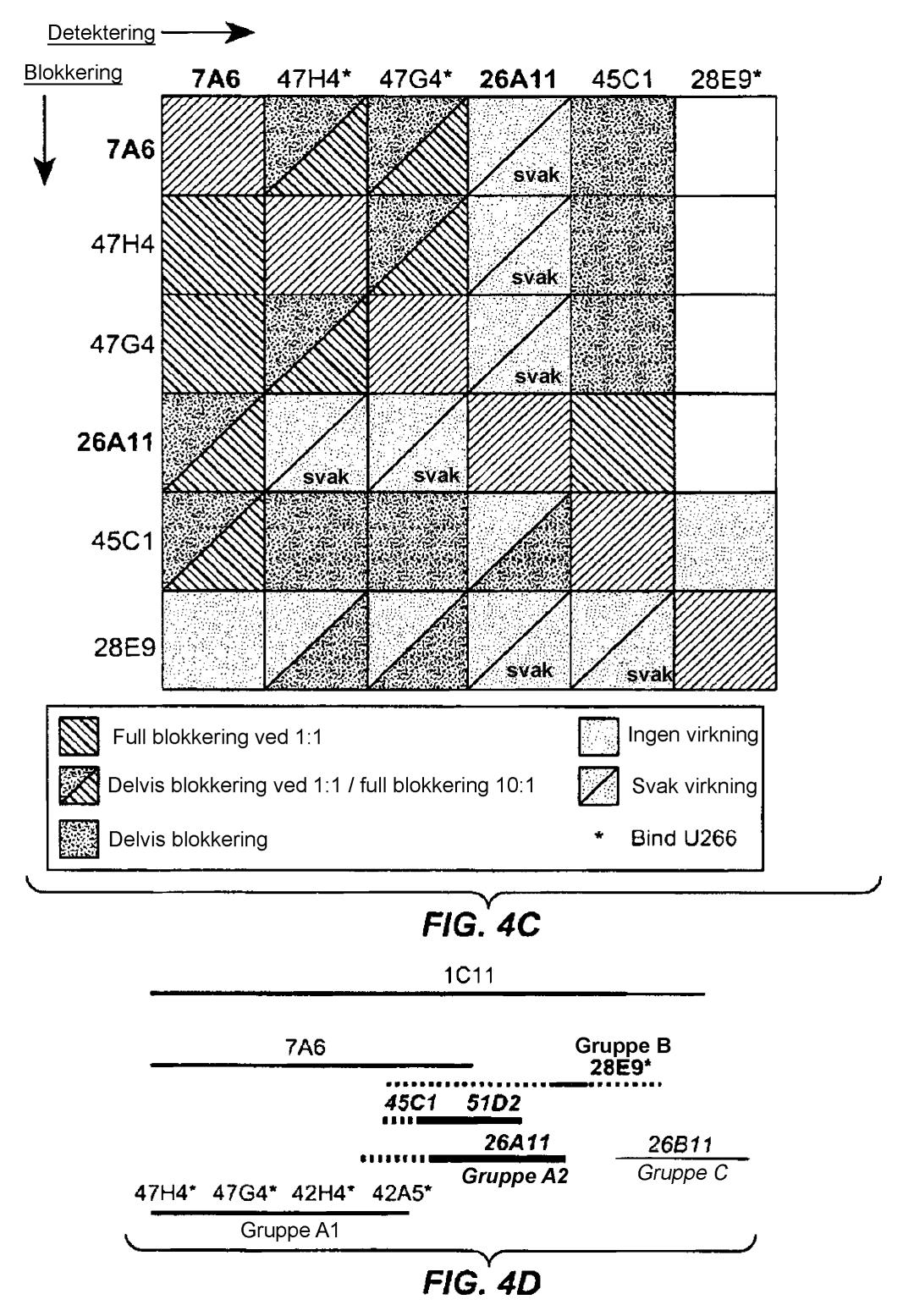
262 39 NO/EP
Søk. Nøkkelinformasjon. Sammendrag og figur. Klasser. IPC-klasse. Søker. Innehaver. Finn patenter, varemerker og design i Norge
 Søk Finn patenter, varemerker og design i Norge Nøkkelinformasjon Databasen er sist oppdatert 2017.09.30 12:04:00 Tittel Status Hovedstatus Detaljstatus Patentnummer Europeisk (EP) publiserings nummer
Søk Finn patenter, varemerker og design i Norge Nøkkelinformasjon Databasen er sist oppdatert 2017.09.30 12:04:00 Tittel Status Hovedstatus Detaljstatus Patentnummer Europeisk (EP) publiserings nummer
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2310382 B1 (19) NO NORGE (51) Int Cl. C07D 401/12 (2006.01) A61K 31/4412 (2006.01) A61P 35/00 (2006.01) C07D 401/14 (2006.01) C07D 403/12 (2006.01)
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2310382 B1 (19) NO NORGE (51) Int Cl. C07D 401/12 (2006.01) A61K 31/4412 (2006.01) A61P 35/00 (2006.01) C07D 401/14 (2006.01) C07D 403/12 (2006.01)
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2011486 B1 (19) NO NORGE (51) Int Cl. A61K 9/20 (2006.01) A61K 31/44 (2006.01) Patentstyret (21) Oversettelse publisert 2012.09.17 (80) Dato for Den
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2011486 B1 (19) NO NORGE (51) Int Cl. A61K 9/20 (2006.01) A61K 31/44 (2006.01) Patentstyret (21) Oversettelse publisert 2012.09.17 (80) Dato for Den
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2272978 B1 (19) NO NORGE (51) Int Cl. C12Q 1/68 (2006.01) Patentstyret (21) Oversettelse publisert 2012.08.13 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2272978 B1 (19) NO NORGE (51) Int Cl. C12Q 1/68 (2006.01) Patentstyret (21) Oversettelse publisert 2012.08.13 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2148670 B1 (19) NO NORGE (51) Int Cl. A61K 31/137 (2006.01) A61P 25/04 (2006.01) Patentstyret (21) Oversettelse publisert 2012.04.02 (80) Dato for
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2148670 B1 (19) NO NORGE (51) Int Cl. A61K 31/137 (2006.01) A61P 25/04 (2006.01) Patentstyret (21) Oversettelse publisert 2012.04.02 (80) Dato for
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2178851 B1 (19) NO NORGE (51) Int Cl. C07D 261/08 (2006.01) A61K 31/42 (2006.01) A61P 3/06 (2006.01) C07D 413/12 (2006.01) Patentstyret (21) Oversettelse
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2178851 B1 (19) NO NORGE (51) Int Cl. C07D 261/08 (2006.01) A61K 31/42 (2006.01) A61P 3/06 (2006.01) C07D 413/12 (2006.01) Patentstyret (21) Oversettelse
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2129377 B1 (19) NO NORGE (51) Int Cl. A61K 31/451 (2006.01) A61K 9/08 (2006.01) A61P 25/00 (2006.01) Patentstyret (21) Oversettelse publisert 2012.01.23
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2129377 B1 (19) NO NORGE (51) Int Cl. A61K 31/451 (2006.01) A61K 9/08 (2006.01) A61P 25/00 (2006.01) Patentstyret (21) Oversettelse publisert 2012.01.23
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2114970 B1 (19) NO NORGE (51) Int Cl. C07F 9/58 (2006.01) A61K 31/44 (2006.01) A61P 1/00 (2006.01) A61P 11/06 (2006.01) A61P 19/02 (2006.01) A61P
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2114970 B1 (19) NO NORGE (51) Int Cl. C07F 9/58 (2006.01) A61K 31/44 (2006.01) A61P 1/00 (2006.01) A61P 11/06 (2006.01) A61P 19/02 (2006.01) A61P
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2246321 B1 (19) NO NORGE (51) Int Cl. A61K 9/20 (2006.01) A61K 31/135 (2006.01) C07C 211/42 (2006.01) Patentstyret (21) Oversettelse publisert 2011.12.12
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2246321 B1 (19) NO NORGE (51) Int Cl. A61K 9/20 (2006.01) A61K 31/135 (2006.01) C07C 211/42 (2006.01) Patentstyret (21) Oversettelse publisert 2011.12.12
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") NO/EP22342 (12) Oversettelse av europeisk patentskrift (11) NO/EP 22342 B1 (19) NO NORGE (1) Int Cl. F2D 23/04 (06.01) Patentstyret (21) Oversettelse publisert 14.01.27 (80) Dato for Den Europeiske Patentmyndighets
NO/EP22342 (12) Oversettelse av europeisk patentskrift (11) NO/EP 22342 B1 (19) NO NORGE (1) Int Cl. F2D 23/04 (06.01) Patentstyret (21) Oversettelse publisert 14.01.27 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2170890 B1 (19) NO NORGE (51) Int Cl. C07D 487/04 (2006.01) Patentstyret (21) Oversettelse publisert 2012.03.12 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2170890 B1 (19) NO NORGE (51) Int Cl. C07D 487/04 (2006.01) Patentstyret (21) Oversettelse publisert 2012.03.12 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2274977 B1 (19) NO NORGE (1) Int Cl. A01K 83/00 (2006.01) Patentstyret (21) Oversettelse publisert 2014.02.17 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2274977 B1 (19) NO NORGE (1) Int Cl. A01K 83/00 (2006.01) Patentstyret (21) Oversettelse publisert 2014.02.17 (80) Dato for Den Europeiske Patentmyndighets
europeisk patentskrift
 (12) Oversettelse av europeisk patentskrift (11) NO/EP 2384729 B1 (19) NO NORGE (1) Int Cl. A61G /12 (2006.01) Patentstyret (21) Oversettelse publisert 2013.04.08 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2384729 B1 (19) NO NORGE (1) Int Cl. A61G /12 (2006.01) Patentstyret (21) Oversettelse publisert 2013.04.08 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 240726 B1 (19) NO NORGE (1) Int Cl. H0K 3/36 (2006.01) H0K 3/42 (2006.01) H0K 3/46 (2006.01) Patentstyret (21) Oversettelse publisert 2014.03.17 (80)
(12) Oversettelse av europeisk patentskrift (11) NO/EP 240726 B1 (19) NO NORGE (1) Int Cl. H0K 3/36 (2006.01) H0K 3/42 (2006.01) H0K 3/46 (2006.01) Patentstyret (21) Oversettelse publisert 2014.03.17 (80)
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2252286 B1 (19) NO NORGE (51) Int Cl. A61K 31/357 (2006.01) Patentstyret (21) Oversettelse publisert 2012.01.16 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2252286 B1 (19) NO NORGE (51) Int Cl. A61K 31/357 (2006.01) Patentstyret (21) Oversettelse publisert 2012.01.16 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") 1 3 (12) Oversettelse av europeisk patentskrift (11) NO/EP 2207775 B1 (19) NO NORGE (51) Int Cl. C07D 401/12 (2006.01) A61K 31/5377 (2006.01) A61P 3/06 (2006.01) C07D 401/14 (2006.01) C07D 413/14 (2006.01)
1 3 (12) Oversettelse av europeisk patentskrift (11) NO/EP 2207775 B1 (19) NO NORGE (51) Int Cl. C07D 401/12 (2006.01) A61K 31/5377 (2006.01) A61P 3/06 (2006.01) C07D 401/14 (2006.01) C07D 413/14 (2006.01)
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2128505 B1 (19) NO NORGE (51) Int Cl. F16L 9/12 (2006.01) F16L 3/14 (2006.01) F16L 11/127 (2006.01) F24F 13/02 (2006.01) H05F 3/02 (2006.01) Patentstyret
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2128505 B1 (19) NO NORGE (51) Int Cl. F16L 9/12 (2006.01) F16L 3/14 (2006.01) F16L 11/127 (2006.01) F24F 13/02 (2006.01) H05F 3/02 (2006.01) Patentstyret
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 213696 B1 (19) NO NORGE (1) Int Cl. B23K 9/32 (2006.01) B23K 9/28 (2006.01) Patentstyret (21) Oversettelse publisert 2014.04.07 (80) Dato for Den
(12) Oversettelse av europeisk patentskrift (11) NO/EP 213696 B1 (19) NO NORGE (1) Int Cl. B23K 9/32 (2006.01) B23K 9/28 (2006.01) Patentstyret (21) Oversettelse publisert 2014.04.07 (80) Dato for Den
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 270722 B1 (19) NO NORGE (1) Int Cl. F21V 23/02 (06.01) F21S 8/02 (06.01) F21V 23/00 (06.01) Patentstyret (21) Oversettelse publisert 14.03. (80) Dato
(12) Oversettelse av europeisk patentskrift (11) NO/EP 270722 B1 (19) NO NORGE (1) Int Cl. F21V 23/02 (06.01) F21S 8/02 (06.01) F21V 23/00 (06.01) Patentstyret (21) Oversettelse publisert 14.03. (80) Dato
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2285808 B1 (19) NO NORGE (51) Int Cl. C07D 471/20 (2006.01) A61K 31/407 (2006.01) A61K 31/424 (2006.01) A61K 31/437 (2006.01) A61K 31/438 (2006.01)
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2285808 B1 (19) NO NORGE (51) Int Cl. C07D 471/20 (2006.01) A61K 31/407 (2006.01) A61K 31/424 (2006.01) A61K 31/437 (2006.01) A61K 31/438 (2006.01)
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 218466 B1 (19) NO NORGE (1) Int Cl. B67C 3/26 (06.01) B6D 47/ (06.01) B67C 7/00 (06.01) Patentstyret (21) Oversettelse publisert 12.02. (80) Dato
(12) Oversettelse av europeisk patentskrift (11) NO/EP 218466 B1 (19) NO NORGE (1) Int Cl. B67C 3/26 (06.01) B6D 47/ (06.01) B67C 7/00 (06.01) Patentstyret (21) Oversettelse publisert 12.02. (80) Dato
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 88493 B1 (19) NO NORGE (1) Int Cl. G06F 1/00 (06.01) H01L 23/34 (06.01) G06F 1/ (06.01) Patentstyret (21) Oversettelse publisert 13.04.22 (80) Dato
(12) Oversettelse av europeisk patentskrift (11) NO/EP 88493 B1 (19) NO NORGE (1) Int Cl. G06F 1/00 (06.01) H01L 23/34 (06.01) G06F 1/ (06.01) Patentstyret (21) Oversettelse publisert 13.04.22 (80) Dato
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2613860 B1 (19) NO NORGE (51) Int Cl. B01D 15/18 (2006.01) C11B 3/10 (2006.01) C11C 1/00 (2006.01) C11C 1/08 (2006.01) Patentstyret (21) Oversettelse
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2613860 B1 (19) NO NORGE (51) Int Cl. B01D 15/18 (2006.01) C11B 3/10 (2006.01) C11C 1/00 (2006.01) C11C 1/08 (2006.01) Patentstyret (21) Oversettelse
europeisk patentskrift
 (12) Oversettelse av europeisk patentskrift (11) NO/EP 2125711 B1 (19) NO NORGE (51) Int Cl. C07C 321/20 (2006.01) A61K 31/216 (2006.01) A61K 31/421 (2006.01) A61K 31/4402 (2006.01) A61K 31/495 (2006.01)
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2125711 B1 (19) NO NORGE (51) Int Cl. C07C 321/20 (2006.01) A61K 31/216 (2006.01) A61K 31/421 (2006.01) A61K 31/4402 (2006.01) A61K 31/495 (2006.01)
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 237066 B1 (19) NO NORGE (1) Int Cl. E06C 1/12 (06.01) Patentstyret (21) Oversettelse publisert 14.02.24 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 237066 B1 (19) NO NORGE (1) Int Cl. E06C 1/12 (06.01) Patentstyret (21) Oversettelse publisert 14.02.24 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2173868 B1 (19) NO NORGE (51) Int Cl. C12N 9/50 (2006.01) C07K 14/415 (2006.01) C12N 15/29 (2006.01) C12N 15/57 (2006.01) C12N 15/81 (2006.01) A23J
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2173868 B1 (19) NO NORGE (51) Int Cl. C12N 9/50 (2006.01) C07K 14/415 (2006.01) C12N 15/29 (2006.01) C12N 15/57 (2006.01) C12N 15/81 (2006.01) A23J
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2445326 B1 (19) NO NORGE (51) Int Cl. H05K 5/02 (2006.01) B43K 23/12 (2006.01) B43K 24/06 (2006.01) H01R 13/60 (2006.01) Patentstyret (21) Oversettelse
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2445326 B1 (19) NO NORGE (51) Int Cl. H05K 5/02 (2006.01) B43K 23/12 (2006.01) B43K 24/06 (2006.01) H01R 13/60 (2006.01) Patentstyret (21) Oversettelse
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2216387 B1 (19) NO NORGE (51) Int Cl. C10L 5/44 (2006.01) C10L 5/14 (2006.01) C10L 5/36 (2006.01) Patentstyret (21) Oversettelse publisert 2013.05.06
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2216387 B1 (19) NO NORGE (51) Int Cl. C10L 5/44 (2006.01) C10L 5/14 (2006.01) C10L 5/36 (2006.01) Patentstyret (21) Oversettelse publisert 2013.05.06
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 223094 B1 (19) NO NORGE (1) Int Cl. A43B 7/32 (06.01) A43B 7/12 (06.01) A43B 7/34 (06.01) A43B 13/12 (06.01) A43B 13/41 (06.01) B29D 3/14 (.01) Patentstyret
(12) Oversettelse av europeisk patentskrift (11) NO/EP 223094 B1 (19) NO NORGE (1) Int Cl. A43B 7/32 (06.01) A43B 7/12 (06.01) A43B 7/34 (06.01) A43B 13/12 (06.01) A43B 13/41 (06.01) B29D 3/14 (.01) Patentstyret
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") NO/EP2770 (12) Oversettelse av europeisk patentskrift (11) NO/EP 2770 B1 (19) NO NORGE (1) Int Cl. B23K 3/00 (06.01) C21D 6/00 (06.01) C21D 9/04 (06.01) C22C 38/00 (06.01) C22C 38/44 (06.01) Patentstyret
NO/EP2770 (12) Oversettelse av europeisk patentskrift (11) NO/EP 2770 B1 (19) NO NORGE (1) Int Cl. B23K 3/00 (06.01) C21D 6/00 (06.01) C21D 9/04 (06.01) C22C 38/00 (06.01) C22C 38/44 (06.01) Patentstyret
europeisk patentskrift
 (12) Oversettelse av europeisk patentskrift (11) NO/EP 2184425 B1 (19) NO NORGE (51) Int Cl. E05B 17/20 (2006.01) E05B 63/00 (2006.01) Patentstyret (21) Oversettelse publisert 2012.02.06 (80) Dato for
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2184425 B1 (19) NO NORGE (51) Int Cl. E05B 17/20 (2006.01) E05B 63/00 (2006.01) Patentstyret (21) Oversettelse publisert 2012.02.06 (80) Dato for
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 224294 B1 (19) NO NORGE (1) Int Cl. F16K 31/44 (2006.01) Patentstyret (21) Oversettelse publisert 2012.04.10 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 224294 B1 (19) NO NORGE (1) Int Cl. F16K 31/44 (2006.01) Patentstyret (21) Oversettelse publisert 2012.04.10 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2491293 B1 (19) NO NORGE (1) Int Cl. F17C 3/02 (06.01) Patentstyret (21) Oversettelse publisert 13.11.2 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2491293 B1 (19) NO NORGE (1) Int Cl. F17C 3/02 (06.01) Patentstyret (21) Oversettelse publisert 13.11.2 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 24012 B1 (19) NO NORGE (1) Int Cl. B2C 1/00 (2006.01) B2C 1/06 (2006.01) Patentstyret (21) Oversettelse publisert 2014.12.22 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift (11) NO/EP 24012 B1 (19) NO NORGE (1) Int Cl. B2C 1/00 (2006.01) B2C 1/06 (2006.01) Patentstyret (21) Oversettelse publisert 2014.12.22 (80) Dato for Den Europeiske
Forløp av ikke-adaptiv og adaptiv immunrespons. Mononukleære celler, metylfiolett farging
 Forløp av ikke-adaptiv og adaptiv immunrespons Mononukleære celler, metylfiolett farging 1 Nøytrofile granulocytter Gjenkjennelsesprinsipper medfødt vs. adaptiv immunitet Toll Like Receptors Mikroorganismer
Forløp av ikke-adaptiv og adaptiv immunrespons Mononukleære celler, metylfiolett farging 1 Nøytrofile granulocytter Gjenkjennelsesprinsipper medfødt vs. adaptiv immunitet Toll Like Receptors Mikroorganismer
(12) Oversettelse av europeisk patentskrift. Avviker fra Patent B1 etter innsigelse
 Oversettelse av europeisk patentskrift. Avviker fra Patent B1 etter innsigelse") (12) Oversettelse av europeisk patentskrift (11) NO/EP 217368 B2 (19) NO NORGE (1) Int Cl. B42D / (06.01) Patentstyret Avviker fra Patent B1 etter innsigelse (21) Oversettelse publisert.04. (80) Dato for
(12) Oversettelse av europeisk patentskrift (11) NO/EP 217368 B2 (19) NO NORGE (1) Int Cl. B42D / (06.01) Patentstyret Avviker fra Patent B1 etter innsigelse (21) Oversettelse publisert.04. (80) Dato for
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 11438 B1 (19) NO NORGE (1) Int Cl. E04B 1/343 (06.01) B63B 29/02 (06.01) Patentstyret (21) Oversettelse publisert.02.23 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift (11) NO/EP 11438 B1 (19) NO NORGE (1) Int Cl. E04B 1/343 (06.01) B63B 29/02 (06.01) Patentstyret (21) Oversettelse publisert.02.23 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2317621 B1 (19) NO NORGE (1) Int Cl. H02G 3/12 (06.01) Patentstyret (21) Oversettelse publisert 1.02.02 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2317621 B1 (19) NO NORGE (1) Int Cl. H02G 3/12 (06.01) Patentstyret (21) Oversettelse publisert 1.02.02 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2311023 B1 (19) NO NORGE (51) Int Cl. G09F 17/00 (2006.01) Patentstyret (21) Oversettelse publisert 2014.02.17 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2311023 B1 (19) NO NORGE (51) Int Cl. G09F 17/00 (2006.01) Patentstyret (21) Oversettelse publisert 2014.02.17 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 22619 B1 (19) NO NORGE (1) Int Cl. B21D 1/4 (2006.01) B21K 21/04 (2006.01) F42B /02 (2006.01) F42B /188 (2006.01) Patentstyret (21) Oversettelse publisert
(12) Oversettelse av europeisk patentskrift (11) NO/EP 22619 B1 (19) NO NORGE (1) Int Cl. B21D 1/4 (2006.01) B21K 21/04 (2006.01) F42B /02 (2006.01) F42B /188 (2006.01) Patentstyret (21) Oversettelse publisert
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2082973 B1 (19) NO NORGE (1) Int Cl. B6D 81/34 (2006.01) Patentstyret (21) Oversettelse publisert 2014.06.02 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2082973 B1 (19) NO NORGE (1) Int Cl. B6D 81/34 (2006.01) Patentstyret (21) Oversettelse publisert 2014.06.02 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 22799 B1 (19) NO NORGE (1) Int Cl. A61K 31/23 (06.01) A61K 31/047 (06.01) A61K 31/231 (06.01) A61K 31/232 (06.01) A61K 31/3 (06.01) A61K 31/93 (06.01)
(12) Oversettelse av europeisk patentskrift (11) NO/EP 22799 B1 (19) NO NORGE (1) Int Cl. A61K 31/23 (06.01) A61K 31/047 (06.01) A61K 31/231 (06.01) A61K 31/232 (06.01) A61K 31/3 (06.01) A61K 31/93 (06.01)
APOPTOTISKE ANTISTOFFER MOT IGE SOM BINDER MEMBRANBUNDET IGE
 1 APOPTOTISKE ANTISTOFFER MOT IGE SOM BINDER MEMBRANBUNDET IGE RELASJON TILBAKE I TID Etter 3 USC 119 (e) krever oppfinnelsen prioritet over USSN 60/896,339, arkivert den 22. mars 2007. OPPFINNELSENS BAKGRUNN
1 APOPTOTISKE ANTISTOFFER MOT IGE SOM BINDER MEMBRANBUNDET IGE RELASJON TILBAKE I TID Etter 3 USC 119 (e) krever oppfinnelsen prioritet over USSN 60/896,339, arkivert den 22. mars 2007. OPPFINNELSENS BAKGRUNN
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2636033 B1 (19) NO NORGE (51) Int Cl. Patentstyret G09B 23/28 (2006.01) G09B 23/30 (2006.01) (21) Oversettelse publisert 2015.11.09 (80) Dato for
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2636033 B1 (19) NO NORGE (51) Int Cl. Patentstyret G09B 23/28 (2006.01) G09B 23/30 (2006.01) (21) Oversettelse publisert 2015.11.09 (80) Dato for
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2217383 B1 (19) NO NORGE (1) Int Cl. B0B 12/00 (06.01) B0B 11/00 (06.01) G01F 11/02 (06.01) G01F 1/07 (06.01) G07C 3/04 (06.01) Patentstyret (21)
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2217383 B1 (19) NO NORGE (1) Int Cl. B0B 12/00 (06.01) B0B 11/00 (06.01) G01F 11/02 (06.01) G01F 1/07 (06.01) G07C 3/04 (06.01) Patentstyret (21)
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 242166 B1 (19) NO NORGE (1) Int Cl. G06K 19/077 (06.01) G06K 19/06 (06.01) Patentstyret (21) Oversettelse publisert 14.02.24 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift (11) NO/EP 242166 B1 (19) NO NORGE (1) Int Cl. G06K 19/077 (06.01) G06K 19/06 (06.01) Patentstyret (21) Oversettelse publisert 14.02.24 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2261144 B1 (19) NO NORGE (1) Int Cl. B6G 21/00 (06.01) B6G 21/08 (06.01) Patentstyret (21) Oversettelse publisert 13.07.08 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2261144 B1 (19) NO NORGE (1) Int Cl. B6G 21/00 (06.01) B6G 21/08 (06.01) Patentstyret (21) Oversettelse publisert 13.07.08 (80) Dato for Den Europeiske
europeisk patentskrift
 (12) Oversettelse av europeisk patentskrift (11) NO/EP 2238877 B1 (19) NO NORGE (1) Int Cl. A47J 31/08 (06.01) Patentstyret (21) Oversettelse publisert 13.03.11 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2238877 B1 (19) NO NORGE (1) Int Cl. A47J 31/08 (06.01) Patentstyret (21) Oversettelse publisert 13.03.11 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2146022 B1 (19) NO NORGE (1) Int Cl. E04F /06 (2006.01) Patentstyret (21) Oversettelse publisert 2014.11.03 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2146022 B1 (19) NO NORGE (1) Int Cl. E04F /06 (2006.01) Patentstyret (21) Oversettelse publisert 2014.11.03 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 7044 B1 (19) NO NORGE (1) Int Cl. A61K 36/18 (06.01) A61K 33/04 (06.01) A61K 33/18 (06.01) A61K 33/ (06.01) A61K 36/22 (06.01) A61K 36/28 (06.01)
(12) Oversettelse av europeisk patentskrift (11) NO/EP 7044 B1 (19) NO NORGE (1) Int Cl. A61K 36/18 (06.01) A61K 33/04 (06.01) A61K 33/18 (06.01) A61K 33/ (06.01) A61K 36/22 (06.01) A61K 36/28 (06.01)
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2264391 B1 (19) NO NORGE (1) Int Cl. F27D 3/1 (2006.01) C21B 7/12 (2006.01) Patentstyret (21) Oversettelse publisert 2013.11.18 (80) Dato for Den
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2264391 B1 (19) NO NORGE (1) Int Cl. F27D 3/1 (2006.01) C21B 7/12 (2006.01) Patentstyret (21) Oversettelse publisert 2013.11.18 (80) Dato for Den
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2672278 B1 (19) NO NORGE (1) Int Cl. G01R 1/067 (2006.01) G01R 1/04 (2006.01) G01R 19/1 (2006.01) Patentstyret (21) Oversettelse publisert 201.04.20
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2672278 B1 (19) NO NORGE (1) Int Cl. G01R 1/067 (2006.01) G01R 1/04 (2006.01) G01R 19/1 (2006.01) Patentstyret (21) Oversettelse publisert 201.04.20
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 19724 B1 (19) NO NORGE (1) Int Cl. B63H 23/02 (06.01) Patentstyret (21) Oversettelse publisert 12.12. (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 19724 B1 (19) NO NORGE (1) Int Cl. B63H 23/02 (06.01) Patentstyret (21) Oversettelse publisert 12.12. (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2231428 B1 (19) NO NORGE (1) Int Cl. B60H 1/32 (06.01) Patentstyret (21) Oversettelse publisert 12.11.26 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2231428 B1 (19) NO NORGE (1) Int Cl. B60H 1/32 (06.01) Patentstyret (21) Oversettelse publisert 12.11.26 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift. Avviker fra Patent B1 etter innsigelse
 Oversettelse av europeisk patentskrift. Avviker fra Patent B1 etter innsigelse") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2175588 B2 (19) NO NORGE (51) Int Cl. H04L 12/14 (2006.01) H04L 29/08 (2006.01) Patentstyret Avviker fra Patent B1 etter innsigelse (21) Oversettelse
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2175588 B2 (19) NO NORGE (51) Int Cl. H04L 12/14 (2006.01) H04L 29/08 (2006.01) Patentstyret Avviker fra Patent B1 etter innsigelse (21) Oversettelse
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2236434 B1 (19) NO NORGE (1) Int Cl. B6D 77/04 (06.01) B6D 77/06 (06.01) Patentstyret (21) Oversettelse publisert 11.12.19 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2236434 B1 (19) NO NORGE (1) Int Cl. B6D 77/04 (06.01) B6D 77/06 (06.01) Patentstyret (21) Oversettelse publisert 11.12.19 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2708433 B1 (19) NO NORGE (1) Int Cl. B61B 1/02 (2006.01) B61B 12/02 (2006.01) Patentstyret (21) Oversettelse publisert 201.01.12 (80) Dato for Den
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2708433 B1 (19) NO NORGE (1) Int Cl. B61B 1/02 (2006.01) B61B 12/02 (2006.01) Patentstyret (21) Oversettelse publisert 201.01.12 (80) Dato for Den
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2096736 B1 (19) NO NORGE (1) Int Cl. H02K 1/32 (2006.01) H02K 3/24 (2006.01) H02K 9/00 (2006.01) Patentstyret (21) Oversettelse publisert 2011.09.0
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2096736 B1 (19) NO NORGE (1) Int Cl. H02K 1/32 (2006.01) H02K 3/24 (2006.01) H02K 9/00 (2006.01) Patentstyret (21) Oversettelse publisert 2011.09.0
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2097141 B1 (19) NO NORGE (51) Int Cl. A62B 35/00 (2006.01) Patentstyret (21) Oversettelse publisert 2013.08.19 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2097141 B1 (19) NO NORGE (51) Int Cl. A62B 35/00 (2006.01) Patentstyret (21) Oversettelse publisert 2013.08.19 (80) Dato for Den Europeiske Patentmyndighets
NITO Bioingeniørfaglig Institutt kurs i Immunologi The Edge, Tromsø, 11. februar 2015. Generell Immunologi
 NITO Bioingeniørfaglig Institutt kurs i Immunologi The Edge, Tromsø, 11. februar 2015 Generell Immunologi Tor B Stuge Immunologisk forskningsgruppe, IMB Universitetet i Tromsø Innhold: 1. Immunsystemets
NITO Bioingeniørfaglig Institutt kurs i Immunologi The Edge, Tromsø, 11. februar 2015 Generell Immunologi Tor B Stuge Immunologisk forskningsgruppe, IMB Universitetet i Tromsø Innhold: 1. Immunsystemets
(12) Translation of european patent specification
 Translation of european patent specification") NO/EP20 (12) Translation of european patent specification (11) NO/EP 20 B1 (19) NO NORWAY (1) Int Cl. C07K 16/28 (2006.01) A61P 3/00 (2006.01) A61P 37/00 (2006.01) Norwegian Industrial Property Office
NO/EP20 (12) Translation of european patent specification (11) NO/EP 20 B1 (19) NO NORWAY (1) Int Cl. C07K 16/28 (2006.01) A61P 3/00 (2006.01) A61P 37/00 (2006.01) Norwegian Industrial Property Office
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 240126 B1 (19) NO NORGE (1) Int Cl. C07D 211/62 (06.01) A61K 31/16 (06.01) A61K 31/44 (06.01) A61K 31/0 (06.01) A61K 31/06 (06.01) C07D 7/277 (06.01)
(12) Oversettelse av europeisk patentskrift (11) NO/EP 240126 B1 (19) NO NORGE (1) Int Cl. C07D 211/62 (06.01) A61K 31/16 (06.01) A61K 31/44 (06.01) A61K 31/0 (06.01) A61K 31/06 (06.01) C07D 7/277 (06.01)
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 261673 B1 (19) NO NORGE (1) Int Cl. B60H 1/32 (06.01) B60H 1/00 (06.01) Patentstyret (21) Oversettelse publisert 1.01.12 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift (11) NO/EP 261673 B1 (19) NO NORGE (1) Int Cl. B60H 1/32 (06.01) B60H 1/00 (06.01) Patentstyret (21) Oversettelse publisert 1.01.12 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 22442 B1 (19) NO NORGE (1) Int Cl. G07B 1/00 (11.01) Patentstyret (21) Oversettelse publisert 13..28 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 22442 B1 (19) NO NORGE (1) Int Cl. G07B 1/00 (11.01) Patentstyret (21) Oversettelse publisert 13..28 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2243894 B1 (19) NO NORGE (1) Int Cl. E04F /06 (2006.01) Patentstyret (21) Oversettelse publisert 201.01.26 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2243894 B1 (19) NO NORGE (1) Int Cl. E04F /06 (2006.01) Patentstyret (21) Oversettelse publisert 201.01.26 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2477830 B1 (19) NO NORGE (1) Int Cl. B60K 1/00 (06.01) Patentstyret (21) Oversettelse publisert 13.12.02 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2477830 B1 (19) NO NORGE (1) Int Cl. B60K 1/00 (06.01) Patentstyret (21) Oversettelse publisert 13.12.02 (80) Dato for Den Europeiske Patentmyndighets
1. Medfødt og ervervet immunitet. Karl Schenck, V2015
 1. Medfødt og ervervet immunitet Karl Schenck, V2015 Medfødt og ervervet immunforsvar Antimicrobial peptides «Alltid beredt!» Relativt uspesifikt Må aktiveres Spesifikt Komponenter av medfødt immunitet
1. Medfødt og ervervet immunitet Karl Schenck, V2015 Medfødt og ervervet immunforsvar Antimicrobial peptides «Alltid beredt!» Relativt uspesifikt Må aktiveres Spesifikt Komponenter av medfødt immunitet
(12) Translation of european patent specification
 Translation of european patent specification") (12) Translation of european patent specification (11) NO/EP 26098 B1 (19) NO NORWAY (1) Int Cl. C07K 14/08 (06.01) C12Q 1/70 (06.01) Norwegian Industrial Property Office (21) Translation Published 16..24
(12) Translation of european patent specification (11) NO/EP 26098 B1 (19) NO NORWAY (1) Int Cl. C07K 14/08 (06.01) C12Q 1/70 (06.01) Norwegian Industrial Property Office (21) Translation Published 16..24
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 23196 B1 (19) NO NORGE (1) Int Cl. A01M 7/00 (06.01) Patentstyret (21) Oversettelse publisert 13.08.19 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 23196 B1 (19) NO NORGE (1) Int Cl. A01M 7/00 (06.01) Patentstyret (21) Oversettelse publisert 13.08.19 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2244923 B1 (19) NO NORGE (1) Int Cl. B61K 9/ (2006.01) Patentstyret (21) Oversettelse publisert 2013.09.30 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2244923 B1 (19) NO NORGE (1) Int Cl. B61K 9/ (2006.01) Patentstyret (21) Oversettelse publisert 2013.09.30 (80) Dato for Den Europeiske Patentmyndighets
(12) Translation of european patent specification
 Translation of european patent specification") (12) Translation of european patent specification (11) NO/EP 211778 B1 (19) NO NORWAY (1) Int Cl. A61K 38/16 (06.01) C07K 7/08 (06.01) Norwegian Industrial Property Office (21) Translation Published 1.09.07
(12) Translation of european patent specification (11) NO/EP 211778 B1 (19) NO NORWAY (1) Int Cl. A61K 38/16 (06.01) C07K 7/08 (06.01) Norwegian Industrial Property Office (21) Translation Published 1.09.07
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2292031 B1 (19) NO NORGE (51) Int Cl. H04W 8/26 (2009.01) Patentstyret (21) Oversettelse publisert 2013.03.25 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2292031 B1 (19) NO NORGE (51) Int Cl. H04W 8/26 (2009.01) Patentstyret (21) Oversettelse publisert 2013.03.25 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") NO/EP28769 (12) Oversettelse av europeisk patentskrift (11) NO/EP 28769 B1 (19) NO NORGE (1) Int Cl. F17D 1/18 (06.01) F16L 3/00 (06.01) Patentstyret (21) Oversettelse publisert 1.04. (80) Dato for Den
NO/EP28769 (12) Oversettelse av europeisk patentskrift (11) NO/EP 28769 B1 (19) NO NORGE (1) Int Cl. F17D 1/18 (06.01) F16L 3/00 (06.01) Patentstyret (21) Oversettelse publisert 1.04. (80) Dato for Den
5. T cellers effektorfunksjoner
 Immunologi 3. semester, V2015 5. T cellers effektorfunksjoner Karl Schenck Institutt for oral biologi Noen typer effektor T celler 3 3 IFNg IL-4 IL-17 IL-10 TGFb TGFb IL-4 Secretory IgA Enhance mast cell
Immunologi 3. semester, V2015 5. T cellers effektorfunksjoner Karl Schenck Institutt for oral biologi Noen typer effektor T celler 3 3 IFNg IL-4 IL-17 IL-10 TGFb TGFb IL-4 Secretory IgA Enhance mast cell
europeisk patentskrift
 (12) Oversettelse av europeisk patentskrift (11) NO/EP 21847 B1 (19) NO NORGE (1) Int Cl. F24F 7/08 (06.01) F24F 11/04 (06.01) F24F 12/00 (06.01) Patentstyret (21) Oversettelse publisert 13.12.02 (80)
(12) Oversettelse av europeisk patentskrift (11) NO/EP 21847 B1 (19) NO NORGE (1) Int Cl. F24F 7/08 (06.01) F24F 11/04 (06.01) F24F 12/00 (06.01) Patentstyret (21) Oversettelse publisert 13.12.02 (80)
europeisk patentskrift
 (12) Oversettelse av europeisk patentskrift (11) NO/EP 17118 B1 (19) NO NORGE (1) Int Cl. B60M 1/06 (06.01) B60M 3/04 (06.01) Patentstyret (21) Oversettelse publisert 14.09.29 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift (11) NO/EP 17118 B1 (19) NO NORGE (1) Int Cl. B60M 1/06 (06.01) B60M 3/04 (06.01) Patentstyret (21) Oversettelse publisert 14.09.29 (80) Dato for Den Europeiske
BIOS 1 Biologi
 . Figurer kapittel 10: Menneskets immunsystem Figur s. 281 En oversikt over immunsystemet og viktige celletyper.> Immunsystemet Uspesifikt immunforsvar Spesifikt immunforsvar Ytre forsvar: hindrer mikroorganismer
. Figurer kapittel 10: Menneskets immunsystem Figur s. 281 En oversikt over immunsystemet og viktige celletyper.> Immunsystemet Uspesifikt immunforsvar Spesifikt immunforsvar Ytre forsvar: hindrer mikroorganismer
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2231500 B1 (19) NO NORGE (51) Int Cl. B66F 9/00 (2006.01) B60P 1/02 (2006.01) B60P 3/022 (2006.01) B62B 3/065 (2006.01) B66D 1/00 (2006.01) B66F 9/06
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2231500 B1 (19) NO NORGE (51) Int Cl. B66F 9/00 (2006.01) B60P 1/02 (2006.01) B60P 3/022 (2006.01) B62B 3/065 (2006.01) B66D 1/00 (2006.01) B66F 9/06
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 216340 B1 (19) NO NORGE (1) Int Cl. B60C 11/11 (06.01) B60C 11/03 (06.01) B60C 11/12 (06.01) Patentstyret (21) Oversettelse publisert 12.12.03 (80)
(12) Oversettelse av europeisk patentskrift (11) NO/EP 216340 B1 (19) NO NORGE (1) Int Cl. B60C 11/11 (06.01) B60C 11/03 (06.01) B60C 11/12 (06.01) Patentstyret (21) Oversettelse publisert 12.12.03 (80)
(86) Europeisk innleveringsdag
 Europeisk innleveringsdag") (12) Oversettelse av europeisk patentskrift (11) NO/EP 297978 B1 (19) NO NORGE (1) Int Cl. A41B 9/02 (06.01) Patentstyret (21) Oversettelse publisert 14.03.17 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 297978 B1 (19) NO NORGE (1) Int Cl. A41B 9/02 (06.01) Patentstyret (21) Oversettelse publisert 14.03.17 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") NO/EP918 (12) Oversettelse av europeisk patentskrift (11) NO/EP 918 B1 (19) NO NORGE (1) Int Cl. H02J 7/00 (06.01) Patentstyret (21) Oversettelse publisert 14.02.03 (80) Dato for Den Europeiske Patentmyndighets
NO/EP918 (12) Oversettelse av europeisk patentskrift (11) NO/EP 918 B1 (19) NO NORGE (1) Int Cl. H02J 7/00 (06.01) Patentstyret (21) Oversettelse publisert 14.02.03 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2146836 B1 (19) NO NORGE (1) Int Cl. A47G 9/ (06.01) B26D 3/00 (06.01) B26D 3/28 (06.01) B29C 44/6 (06.01) Patentstyret (21) Oversettelse publisert
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2146836 B1 (19) NO NORGE (1) Int Cl. A47G 9/ (06.01) B26D 3/00 (06.01) B26D 3/28 (06.01) B29C 44/6 (06.01) Patentstyret (21) Oversettelse publisert
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 238426 B1 (19) NO NORGE (1) Int Cl. G01S 1/68 (06.01) B63C 9/32 (06.01) F41B 13/00 (06.01) F41B 1/00 (06.01) Patentstyret (21) Oversettelse publisert
(12) Oversettelse av europeisk patentskrift (11) NO/EP 238426 B1 (19) NO NORGE (1) Int Cl. G01S 1/68 (06.01) B63C 9/32 (06.01) F41B 13/00 (06.01) F41B 1/00 (06.01) Patentstyret (21) Oversettelse publisert
Oppgave: MED1100-3_OPPGAVE2_H16_KONT
 Side 10 av 35 Oppgave: MED1100-3_OPPGAVE2_H16_KONT Del 1: Ola har en arvelig betinget kombinert immundefekt med mangel på både T-celler og B-celler. Ola får derfor gjentatte Hvorfor er Ola beskyttet mot
Side 10 av 35 Oppgave: MED1100-3_OPPGAVE2_H16_KONT Del 1: Ola har en arvelig betinget kombinert immundefekt med mangel på både T-celler og B-celler. Ola får derfor gjentatte Hvorfor er Ola beskyttet mot
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 21181 B1 (19) NO NORGE (1) Int Cl. F16L 2/00 (2006.01) F16L 33/26 (2006.01) H01P 1/04 (2006.01) Patentstyret (21) Oversettelse publisert 2013.10.28
(12) Oversettelse av europeisk patentskrift (11) NO/EP 21181 B1 (19) NO NORGE (1) Int Cl. F16L 2/00 (2006.01) F16L 33/26 (2006.01) H01P 1/04 (2006.01) Patentstyret (21) Oversettelse publisert 2013.10.28
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2012637 B1 NORGE (19) NO (1) Int Cl. A47K 13/00 (2006.01) Patentstyret (4) Oversettelse publisert: 20.08.09 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2012637 B1 NORGE (19) NO (1) Int Cl. A47K 13/00 (2006.01) Patentstyret (4) Oversettelse publisert: 20.08.09 (80) Dato for Den Europeiske Patentmyndighets
Oppgave: MED1100-3_OPPGAVE2_V18_ORD
 Side 15 av 46 Oppgave: MED1100-3_OPPGAVE2_V18_ORD Del 1: Hvilke av de følgende celler uttrykker normalt (i hvilende tilstand) HLA klasse II molekyler hos mennesket? Angi de tre riktigste svarene. Fibroblaster
Side 15 av 46 Oppgave: MED1100-3_OPPGAVE2_V18_ORD Del 1: Hvilke av de følgende celler uttrykker normalt (i hvilende tilstand) HLA klasse II molekyler hos mennesket? Angi de tre riktigste svarene. Fibroblaster
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 203638 B1 (19) NO NORGE (1) Int Cl. A61K 9/00 (2006.01) A61K 31/382 (2006.01) A61K 31/498 (2006.01) A61K 31/3 (2006.01) Patentstyret (21) Oversettelse
(12) Oversettelse av europeisk patentskrift (11) NO/EP 203638 B1 (19) NO NORGE (1) Int Cl. A61K 9/00 (2006.01) A61K 31/382 (2006.01) A61K 31/498 (2006.01) A61K 31/3 (2006.01) Patentstyret (21) Oversettelse
(12) Oversettelse av europeisk patentskrift. Patentstyret Avviker fra Patent B1 etter innsigelse
 Oversettelse av europeisk patentskrift. Patentstyret Avviker fra Patent B1 etter innsigelse") (12) Oversettelse av europeisk patentskrift (11) NO/EP 21736 B2 (19) NO NORGE (1) Int Cl. A41D 19/00 (06.01) A41D 19/01 (06.01) Patentstyret Avviker fra Patent B1 etter innsigelse (21) Oversettelse publisert
(12) Oversettelse av europeisk patentskrift (11) NO/EP 21736 B2 (19) NO NORGE (1) Int Cl. A41D 19/00 (06.01) A41D 19/01 (06.01) Patentstyret Avviker fra Patent B1 etter innsigelse (21) Oversettelse publisert
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2399741 B1 (19) NO NORGE (1) Int Cl. B32B 27/40 (06.01) C08J 7/04 (06.01) C09D 17/04 (06.01) D21H 19/82 (06.01) Patentstyret (21) Oversettelse publisert
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2399741 B1 (19) NO NORGE (1) Int Cl. B32B 27/40 (06.01) C08J 7/04 (06.01) C09D 17/04 (06.01) D21H 19/82 (06.01) Patentstyret (21) Oversettelse publisert
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2148223 B1 (19) NO NORGE (1) Int Cl. G01V 3/ (06.01) G01V 3/24 (06.01) Patentstyret (21) Oversettelse publisert 13.03.04 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2148223 B1 (19) NO NORGE (1) Int Cl. G01V 3/ (06.01) G01V 3/24 (06.01) Patentstyret (21) Oversettelse publisert 13.03.04 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2289870 B1 (19) NO NORGE (51) Int Cl. C07C 231/24 (2006.01) C07C 237/32 (2006.01) Patentstyret (21) Oversettelse publisert 2013.03.11 (80) Dato for
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2289870 B1 (19) NO NORGE (51) Int Cl. C07C 231/24 (2006.01) C07C 237/32 (2006.01) Patentstyret (21) Oversettelse publisert 2013.03.11 (80) Dato for
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 9863 B1 (19) NO NORGE (1) Int Cl. E04B 2/96 (06.01) Patentstyret (21) Oversettelse publisert 13.09.09 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 9863 B1 (19) NO NORGE (1) Int Cl. E04B 2/96 (06.01) Patentstyret (21) Oversettelse publisert 13.09.09 (80) Dato for Den Europeiske Patentmyndighets
(12) Translation of european patent specification
 Translation of european patent specification") (12) Translation of european patent specification (11) NO/EP 2068927 B1 (19) NO NORWAY (51) Int Cl. A61K 39/395 (2006.01) C07K 16/28 (2006.01) Norwegian Industrial Property Office (21) Translation Published
(12) Translation of european patent specification (11) NO/EP 2068927 B1 (19) NO NORWAY (51) Int Cl. A61K 39/395 (2006.01) C07K 16/28 (2006.01) Norwegian Industrial Property Office (21) Translation Published
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 08940 B1 (19) NO NORGE (1) Int Cl. B6D 2/2 (06.01) A47G 19/34 (06.01) B6D 83/06 (06.01) G01F 11/26 (06.01) Patentstyret (21) Oversettelse publisert
(12) Oversettelse av europeisk patentskrift (11) NO/EP 08940 B1 (19) NO NORGE (1) Int Cl. B6D 2/2 (06.01) A47G 19/34 (06.01) B6D 83/06 (06.01) G01F 11/26 (06.01) Patentstyret (21) Oversettelse publisert
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 1974881 B1 (19) NO NORGE (1) Int Cl. B27B 19/00 (06.01) A61B 17/14 (06.01) Patentstyret (21) Oversettelse publisert 14.01.27 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift (11) NO/EP 1974881 B1 (19) NO NORGE (1) Int Cl. B27B 19/00 (06.01) A61B 17/14 (06.01) Patentstyret (21) Oversettelse publisert 14.01.27 (80) Dato for Den Europeiske
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 20789 B1 (19) NO NORGE (1) Int Cl. B61D 1/00 (06.01) B61D 17/ (06.01) B61D 23/00 (06.01) Patentstyret (21) Oversettelse publisert 12.06.04 (80) Dato
(12) Oversettelse av europeisk patentskrift (11) NO/EP 20789 B1 (19) NO NORGE (1) Int Cl. B61D 1/00 (06.01) B61D 17/ (06.01) B61D 23/00 (06.01) Patentstyret (21) Oversettelse publisert 12.06.04 (80) Dato
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2113323 B1 (19) NO NORGE (1) Int Cl. B23B 31/02 (2006.01) B23B 31/20 (2006.01) Patentstyret (21) Oversettelse publisert 2012.11.19 (80) Dato for Den
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2113323 B1 (19) NO NORGE (1) Int Cl. B23B 31/02 (2006.01) B23B 31/20 (2006.01) Patentstyret (21) Oversettelse publisert 2012.11.19 (80) Dato for Den
(12) Translation of european patent specification
 Translation of european patent specification") (12) Translation of european patent specification (11) NO/EP 2188312 B1 (19) NO NORWAY (1) Int Cl. C07K 16/28 (06.01) A61P 3/00 (06.01) Norwegian Industrial Property Office (21) Translation Published.11.02
(12) Translation of european patent specification (11) NO/EP 2188312 B1 (19) NO NORWAY (1) Int Cl. C07K 16/28 (06.01) A61P 3/00 (06.01) Norwegian Industrial Property Office (21) Translation Published.11.02
(12) Oversettelse av europeisk patentskrift
 Oversettelse av europeisk patentskrift") (12) Oversettelse av europeisk patentskrift (11) NO/EP 2117944 B1 (19) NO NORGE (1) Int Cl. B6D 21/02 (2006.01) Patentstyret (21) Oversettelse publisert 2011.09.0 (80) Dato for Den Europeiske Patentmyndighets
(12) Oversettelse av europeisk patentskrift (11) NO/EP 2117944 B1 (19) NO NORGE (1) Int Cl. B6D 21/02 (2006.01) Patentstyret (21) Oversettelse publisert 2011.09.0 (80) Dato for Den Europeiske Patentmyndighets