Introduksjon til hovedtrekkene i MDR- IDVR regelverket. Hva er nytt sammenliknet med eksisterende regelverk?
|
|
|
- Amanda Ervik
- 5 år siden
- Visninger:
Transkript

1 Introduksjon til hovedtrekkene i MDR- IDVR regelverket. Hva er nytt sammenliknet med eksisterende regelverk? Petter Alexander Strømme - medisinsk utstyr, Legemiddelverket
2 26. mai MDR 19 kalender måneder fra nå 26. mai IVDR 43 kalender måneder fra nå
3 om. medisinsk utstyr og Legemiddelverkets rolle & ansvar nye forordninger (EU) status - Hva er nytt sammenliknet med eksisterende regelverk - Hvilke endringer som kommer - Hva må produsenter av medisinsk utstyr må være spesielt oppmerksomme på når endringene skal implementeres frem mot Hvor finnes relevant veiledningsmateriell fra EU overgangsordninger videre prosess nasjonalt Medisinsk utstyr nye forordninger 3
4
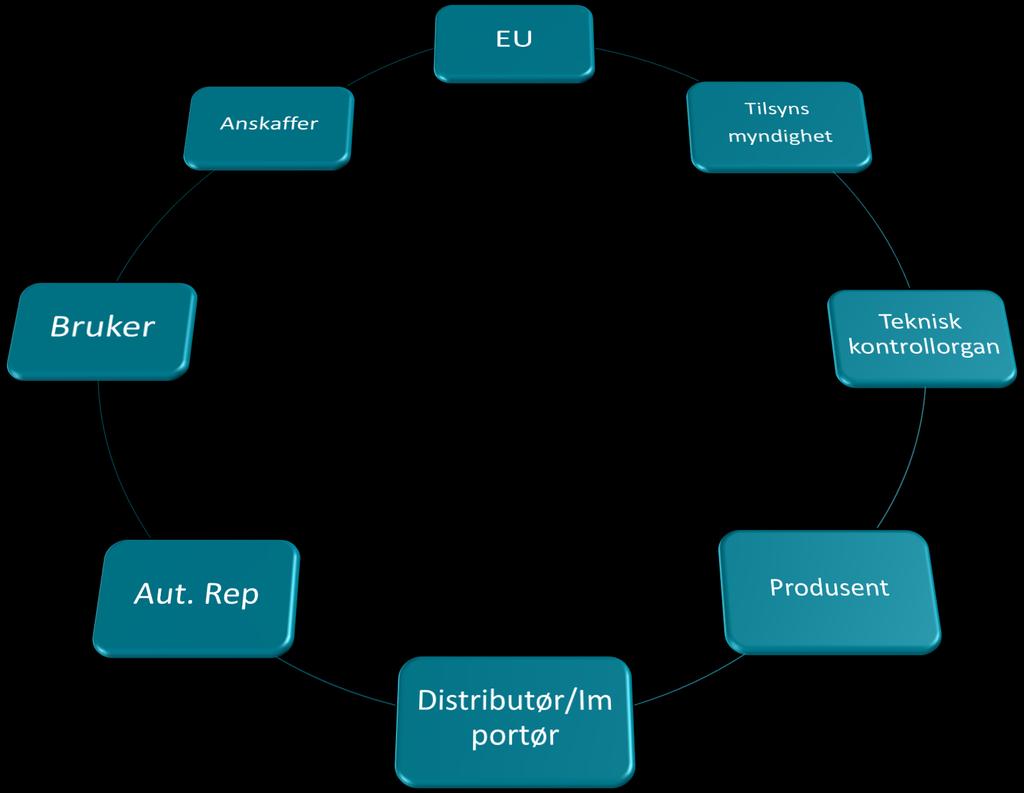

5 Aktører Rolleforståelsen ekstern 5
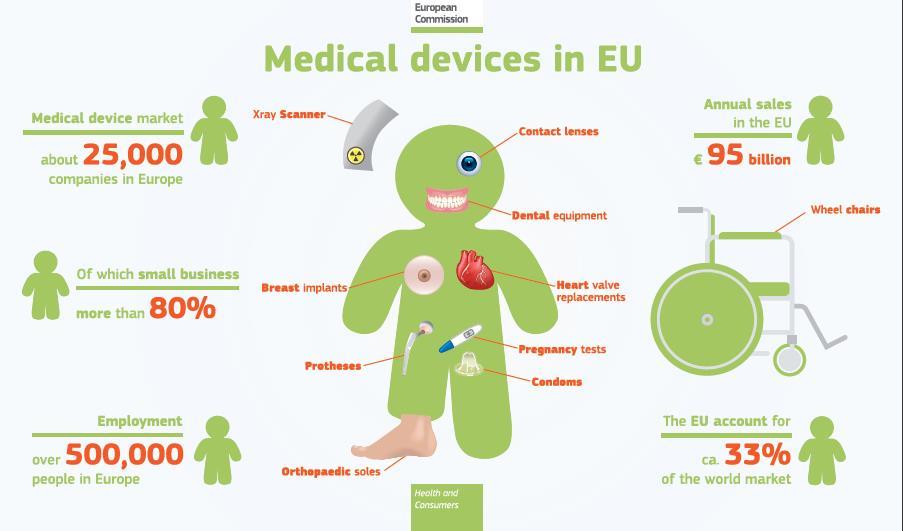
6 6

 Forskrift om håndtering av")
7 Regelverket 90/385: Aktivt implanterbart MU 93/42: Øvrig medisinsk utstyr 98/79: In vitro diagnostisk MU. Lov 12. januar 1995 nr. 6 om medisinsk utstyr Forskrift 15. desember 2005 nr om medisinsk utstyr (nasjonal) Forskrift om håndtering av medisinsk utstyr 7


8 In vitro diagnostisk medisinsk utstyr 8


9 Aktivt implanterbart medisinsk utstyr 9

10 «Øvrig» medisinsk utstyr 10

11 ingen forhåndsgodkjenning av myndigheter til forskjell fra legemidler medisinsk utstyr i høyere risikoklasser vurderes av tekniske kontrollorgan som er utpekt av myndighetene
 Teknisk Klinisk Samsvar med")

 Utstyr på markedet TEKNISK")
12 CE-merking prosess og aktører PRODUSENT (risikoklasse I) Teknisk Klinisk Samsvar med kravene i regelverket? Markedsovervåking Rapportering til myndigheter Klinisk utprøving (øvrige risikoklasser) Utstyr på markedet TEKNISK KONTROLLORGAN MYNDIGHETER Legemiddelverket
13 nye forordninger erstatter dagens regelverk for medisinsk utstyr Directive 90/385/EEC on active implantable medical devices Directive 93/42/EEC on medical devices Regulation on medical devices (MDR) 3 års overgangsperiode Directive 98/79/EC on in vitro diagnostic medical devices Regulation on in vitro diagnostic medical devices (IVDR) 5 års overgangsperiode Entry into force: 25 May 2017 Medisinsk utstyr nye forordninger 13

14


15 Tidslinje : Publisert i Official Journal : Ikrafttredelse /2022: Juridisk gjeldende NB! Bergrepet DOA Date of Application 15

16 Fra direktiver til forordninger. Største regelendring siden 90-tallet Direktivene er over 20 år gamle. Med forordningene er ordlyden på flere vesentlige punkter endret fra direktivene, noe som også har betydning for. Endringene i MDR/IVDR er omfattende og tar høyde for den europeiske utviklingen og som skal sikre et godt og forutsigbart juridisk rammeverk.

17 Medisinsk utstyr nye forordninger DRA-forum
18 + gjennomføringsrettsakter Samtidig er det lagt inn i teksten til forordningene at Kommisjonen skal ha mulighet til å utarbeide opptil 80 rettsakter (både gjennomføringsrettsakter og delegerte rettsakter) hvorav 18 skal være obligatoriske


19 Utvidet definisjon av medisinsk utstyr Utstyr med medisinsk hensikt Medisinsk utstyr nye forordninger DRA-forum
20 Virkeområde NB! Forordningene gjelder for produksjon og omsetting av medisinsk utstyr på det europeiske markedet Fortalen punkt 3 at rettsakten ikke inneholder noen krav til videre bruk av medisinsk utstyr i helsetjenesten. Dette er regler som medlemsstatene selv står fritt til å regulere Forordningene gjelder heller ikke for nasjonal lovgivning om organisering, finansiering av eller tilbud om nasjonale helsetjenester og medisinsk behandling jf. MDR artikkel 1 punkt 15
21 Virkeområde avgrensning andre produkter Forordningen gjelder ikke for legemidler jf. definisjonen i direktiv 2001/83/EF. Det presiseres at legemidler til avansert terapi omfattes av forordning (EF) nr. 1394/2007. Kosmetiske produkter omfattes av forordning (EF) nr. 1223/2009 Mat er regulert i forordning (EF) nr. 178/2002. Gjelder ikke Blod, blodprodukter, plasma og blodceller og dessuten regelverket om organer, vev eller celler av animalsk og human opprinnelse unntatt for medisinsk utstyr som består av inaktiverte organer, celler eller vev. Nytt er at forordningen likevel gjelder for medisinsk utstyr som er produsert ved hjelp av derivater av inaktivert vev eller celler av human opprinnelse. Forordningen er lex specialis til direktiv 2014/30/EU om elektromagnetisk kompatibilitet (EMC-direktivet) samtidig som MDR avgrenser mot direktiv 2006/42/EF om maskiner (maskindirektivet).
22 Nytt virkeområde - Annex XVI Nytt er at forordningens artikkel 1 punkt 2 fastsetter regler om utstyr som har et estetisk og ikke-medisinsk formål. Utstyr som har like egenskaper og en tilsvarende risikoprofil som øvrig medisinsk utstyr. Forordningen presiserer at utstyr med et medisinsk formål og utstyr med et ikke- medisinsk formål skal oppfylle de samme sikkerhetskravene i regelverket. Forordningen vedlegg XVI lister de aktuelle produktkategoriene (derav betegnelsen vedlegg XVI-utstyr):

23 Omfang Kontaktlinser eller annet utstyr som er ment å legges på øyet, for eksempel ikke- korrigerende kontaktlinser med farge Invasivt utstyr som helt eller delvis insettes i kroppen ved hjelp av et kirurgisk inngrep for å endre anatomien, unntatt tatoveringsprodukter og piercing Stoffer for ansiktsbehandling eller annen behandling av hud,- eller slimhinne som injiseres subkutant, submukosalt eller intradermalt, unntatt stoffer til tatovering Utstyr for å redusere og fjerne fettvev, f.eks. utstyr til fettspalting/fettsuging eller utstyr til lipoplasti Utstyr som sender ut elektromagnetisk stråling med høy intensitet (f.eks. infrarødt og ultrafiolett lys) for bruk på menneskekroppen. Det kan være lasere og IPL (intense pulsed light) -utstyr for hudbehandling, fjerning av tatoveringer eller hår. Utstyr til hjernestimulering, dvs. utstyr på elektrisk strøm eller som genererer magnetiske og elektromagnetiske felt som trenger inn i kraniet for å kunne endre den neuronale aktiviteten i hjernen. Medisinsk utstyr nye forordninger 23
24 Annex XVI Forordningen artikkel 1 punkt 2 fastsetter at vedlegg XVIutstyr skal følge visse felles spesifikasjoner (common specifications) som skal fastsettes av Kommisjonen innen 26. mai De felles spesifikasjonene skal minst inneholde regler om risikostyring etter forordningens vedlegg I og nødvendig klinisk utprøving og klinisk evaluering av sikkerheten til slikt utstyr.
25 Definisjoner Definisjonene i MDR og IVDR er betydelig utvidet sammenlignet med direktivene om medisinsk utstyr. Flere av definisjonene samsvarer nå med veletablert praksis i annet produktregelverk for eksempel forordning (EF) nr. 765/2008 (varepakken) som er gjennomført i EØSvareloven. Uttømmende liste over definisjonene i MDR og IVDR følger av artikkel 2 i forordningene. Eksempler på nye definisjoner er: "importør, distributør, medisinsk engangsutstyr, nanomateriale, forfalsket medisinsk utstyr mv. For IVDR er også tester som brukes til målrettet behandling/ å fastslå pasientens egnethet til spesifikk behandling med et legemiddel (companion diagnostics) definert i IVDR.
26 Placing on the market Stilles tydeligere krav til markedsaktører, herunder produsent, importør, distributør og autorisert representant. Nye regler om reklame for medisinsk utstyr - forbud mot villedende reklame. Eksplisitt sagt at utstyr solgt via internett nå også omfattes Kommisjonen kan vedta felles spesifikasjoner (krav til sikkerhet og ytelse). Gjelder nå kun for IVD-direktivet, men muligheten utvides til alt annet medisinsk utstyr. Parallellhandel er tillatt forutsatt at særskilte krav er oppfylt. Uten å få produsentansvar kan importøren/distributøren oversette bruksanvisningen og endre på den ytre emballasjen. Produsenten skal utlevere et implantatkort sammen med implanterbart medisinsk utstyr). Kortet identifiserer utstyret og inneholder nødvendige advarsler/forholdsregler, herunder utstyrets forventede levetid. Egen mal for samsvarserklæring (Declaration of Conformity) jf. vedlegg IV og utforming av EC-sertifikat
27 In-house /reprosessering av engangsutstyr Presiserte krav til produksjon og bruk internt i én helseinstitusjon ("in-house produksjon og bruk). Helseinstitusjoner skal kunne produsere og bruke utstyr internt for å dekke et spesifikt behov til en pasient/pasientgruppe forutsatt at dette ikke gjøres i industriell målestokk og utstyret ikke overføres til en annen juridisk enhet mv. Medlemsstatene kan stille egne krav til reprosessering av medisinsk engangsutstyr. Virksomheter som reprosesserer medisinsk engangsutstyr anses som ny produsent. For reprosessering internt i en helseinstitusjon kan medlemsstatene stille lempligere krav forutsatt at gjenbruken følger felles spesifikasjoner (krav til risikostyring, validering, kvalitetssystem, prosedyrer for å rapportere hendelser og sporbarhet)

28 For produsenter Medisinsk utstyr nye forordninger 28
29 Produsenten skal ha et system for risikostyring Produsenten skal etablere, dokumentere, implementere og vedlikeholde et system for risikostyring som er beskrevet i MDR/IVDR vedlegg I punkt 3. I tillegg skal produsenten utarbeide den tekniske dokumentasjonen som følger av vedlegg II og III. Den tekniske dokumentasjonen omfatter beskrivelse av utstyrets spesifikasjoner, opplysninger om design og produksjon, sikkerhet og ytelse, analyse av forholdet mellom fordeler og risiko og produktverifikasjon (kliniske data, herunder den kliniske evalueringsrapport). Produsenten skal oppbevare den tekniske dokumentasjonen (inkludert samsvarserklæring og sertifikat for utstyr i aktuell risikoklasse) i minst 10 år. For implanterbart utstyr skal dokumentasjonen oppbevares i minst 15 år. Dokumentasjonen skal på anmodning kunne stilles til disposisjon for myndighetene.
30 Kvalitetssystem Produsenten er forpliktet til å etablere et eget kvalitetssystem for sin virksomhet. Kvalitetsstyringssystemet skal dekke alle delene av produsentens virksomhet, dvs. styring, struktur, utvikling av prosedyrer, allokering av ressurser etc. som er nødvendig for å kunne oppfylle kravene i regelverket. Kvalitetsstyringssystem skal oppfylle kravene listet i MDR artikkel 10 punkt 9 og IVDR artikkel 10 punkt 8. Som en del av kvalitetsstyringssystemet skal produsenten ha et system for melding om uønskede hendelser. Dersom produsenten har grunn til å tro at utstyret som omsettes ikke oppfyller sikkerhetskravene skal de rette opp i feilen, evt. tilbakekalle eller trekke utstyret fra markedet. Dersom feilen med utstyret utgjør en alvorlig risiko skal myndighetene varsles.
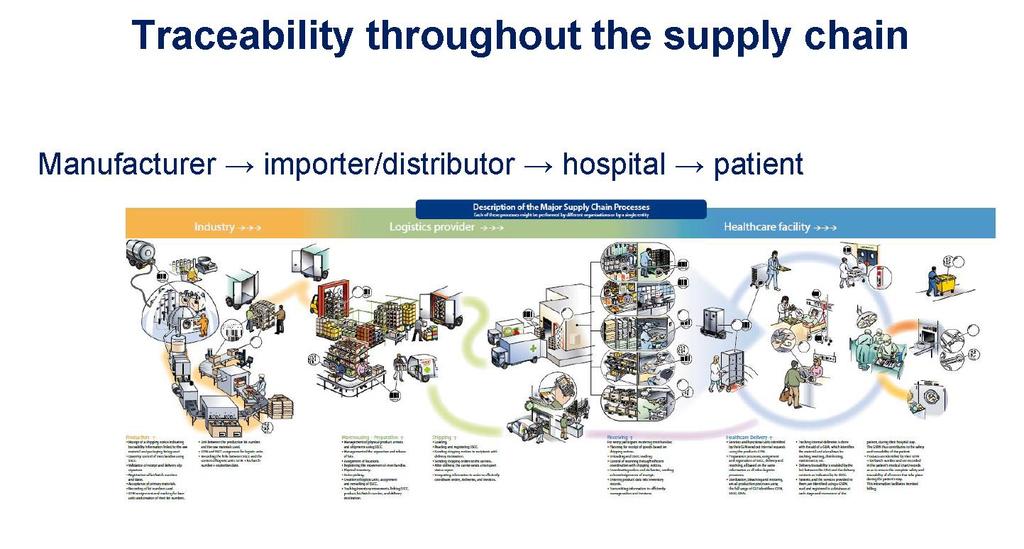
31 Produsenter krav om kompetanse Produsenten skal ha en kvalifisert person internt i organisasjonen som er ansvarlig for «compliance». Bakgrunn fra naturvitenskap, medisin, farmasi, ingeniørfag eller juss. 4 års regulatorisk erfaring eller erfaring med kvalitetsstyringssystem for medisinsk utstyr. «Micro» og små bedrifter er ikke pålagt samme, men skal ha en slik person permanent og kontinuerlig til disposisjon. (Kommisjonsrekommandasjon 2003/362/EF) Medisinsk utstyr nye forordninger 31
32 Produsenter Produsenten skal ha tilstrekkelig økonomisk dekning ved krav om erstatning for skader forårsaket av utstyret. Produsenter er ansvarlig etter produktansvarsloven Medisinsk utstyr nye forordninger 32
33 Autorisert representant Nytt i MDR er at utpekingen av autorisert representant skjer ved fullmakt og at representanten må bekrefte dette skriftlig for at utpekingen skal være gyldig. På forespørsel fra myndighetene skal autorisert representant kunne gi dem en kopi av fullmakten Det er viktig å merke seg at autorisert representant kan bli solidarisk ansvarlig for defekt utstyr på lik linje som produsenten etter MDR art. 11 punkt 6 og IVDR art. 11 punkt 5. Utskiftninger av autorisert representant skal være nærmere fastsatt i en avtale med produsenten jf. MDR/IVDR artikkel 12. Om mulig både med den avgående og tiltredende representanten.
34 Importør 1) Utstyret er CE-merket og at det er utarbeidet en samsvarserklæring 2) Produsenten er klart identifisert og har utpekt en autorisert representant 3) Utstyret er riktig merket og at det følger med en bruksanvisning 4) Produsenten har merket utstyret med en UDI-kode (sporbarhet) 5) Importøren skal på utstyret/emballasjen eller i et dokument som følger utstyret oppgi sitt registrerte firmanavn, registrerte firmaadresse og nærmere kontaktinformasjon. 6) Kontrollere at utstyret er registrert etter MDR artikkel 29 og IVDR artikkel 28 7) Utstyret oppbevares, transporteres og lagres forsvarlig etter bruksanvisningen 8) Det føres et register over klager, utstyr trukket tilbake og gir nødvendig informasjon til produsent, autorisert representant eller distributør. Medisinsk utstyr nye forordninger 34
35 Importør og oppgaver til distributør I tillegg skal importøren: 9) Varsle produsent og autorisert representant dersom importøren har grunn til å tro at det omsettes ulovlig utstyr på markedet. Alvorlig risiko med utstyr skal straks meldes til myndighetene og teknisk kontrollorgan. 10) Oppbevare kopi av samsvarserklæring og evt. sertifikat i 10 år (15 år for implanterbart utstyr). Distributør Distributøren skal som, importøren, oppfylle kravene i punkt 1, 3, 4, 7, 8 og 9. I tillegg skal distributøren sikre at importørens firmanavn, registrerte firmaadresse og nærmere kontaktinformasjon er gjengitt på utstyret/emballasjen eller i et følgedokument. Medisinsk utstyr nye forordninger 35
36 Klassifisering Nytt på IVD A: Lav individuell risiko og lav folkehelse-risiko B: Moderat individuell risiko og/eller lav folkehelse-risiko C: høy individuell risiko og/eller moderat folkehelse-risiko D: høy individuell risiko og høy folkehelse-risiko Nytt: Uenighet mellom produsent og teknisk kontrollorgan mht. klassifisering skal avgjøres av myndighet i landet der produsent holder til og avgjørelsen skal gjøres kjent for medlemsstatene og Kommisjonen.
37 Klassifisering 22 regler for klassifisering er gitt i MDR vedlegg VIII sammenlignet med 18 for MDD F.eks. at: Regel 11 er en ny regel om software; presiserer at software som er medisinsk utstyr kan falle i alle risikoklasser fra I til III, avhengig av bruksområde og risiko. Regel 19 klassifiserer medisinsk utstyr med innhold av nanomaterialer basert på risiko for indre eksponering (IIa, IIb eller III). Regel 20 definerer utstyr til inhalasjon av legemidler til å være risikoklasse IIa eller IIb
38 Klinisk evaluering Forordningen setter detaljerte krav til planlegging, innhold og metodikk for den kliniske evalueringen Produsenten skal dokumentere den kliniske evalueringen i en klinisk evalueringsrapporten. Produsenten pro-aktivt og kontinuerlig skal oppdatere den kliniske evalueringen etter at utstyret er satt på markedet (artikkel 61 og vedlegg XIV). Medisinsk utstyr nye forordninger 38
39 Klinisk evaluering Produsenten gis mulighet til å konsultere kommisjonens ekspertpanel (III, IIb med høy risiko) Sammenliknet med dagens regelverk setter forordningen tydeligere krav dersom klinisk evaluering skal baseres på ekvivalent utstyr fremfor klinisk utprøving. For utstyr uten medisinsk hensikt (vedlegg XVI) skal det foreligge klinisk evaluering hvor utstyrets ytelse og sikkerhet skal demonstreres. For slikt utstyr skal det utføres klinisk utprøving med mindre eksisterende kliniske data fra tilsvarende utstyr kan benyttes Medisinsk utstyr nye forordninger 39
40 Klinisk utprøving Nytt for kliniske utprøvinger i mer enn én medlemsstat Sponsor kan sende inn én søknad og foreslå én koordinerende MS Koordinerende MS skal gi en samlet vurdering av søknaden (assessment-rapport), MS skal vurdere utprøvere og utprøvingssteder, vurdering fra etisk komite, forsikring, samtykkedokumenter. Beslutning fra koordinerende MS skal gjelde, men øvrige MS kan si seg uenige (f.eks. hvis etisk komite i det landet ikke godkjenner utprøvingen) Krav til samtykke generelt, hos personer som ikke kan gi samtykke, mindreårige, nødsituasjoner etc. Medisinsk utstyr nye forordninger 40
41 Sporbarhet Hovedregel: Markedsaktørene skal kunne spore utstyret. Hvem de har fått utstyret fra (f.eks. fra produsent til distributør). Hvem de leverer utstyret til (f.eks. fra distributør til sykehus). Medisinsk utstyr nye forordninger 41
42
43 Sporbarhet - hensyn Hovedhensynene for sporbarhet er: Forbedre systemet for meldinger om uønskede hendelser Legge til rette for effektivt tilsyn fra tilsynsmyndighetene Minske feil i helsetjeneste Hindre forfalskning av medisinsk utstyr Pasientsikkerhet og smittevern

44 SPORBARHET AV MEDISINSK UTSTYR
45 UDI UDI-systemet omfatter følgende elementer: UDI-kode som består av: - Entydig utstyrsidentifikasjonskode (UDI-DI) som er spesifikk for produsenten og et utstyr - Produksjonsidentifikasjonskode (UDI-PI) som identifiserer produksjonen av utstyrsenheten og hvis det er relevant det ferdigpakkede utstyret - UDI-koden skal påføres selve utstyret eller på emballasjen Lagring av UDI-koden av markedsaktører, helseinstitusjoner og helsepersonell
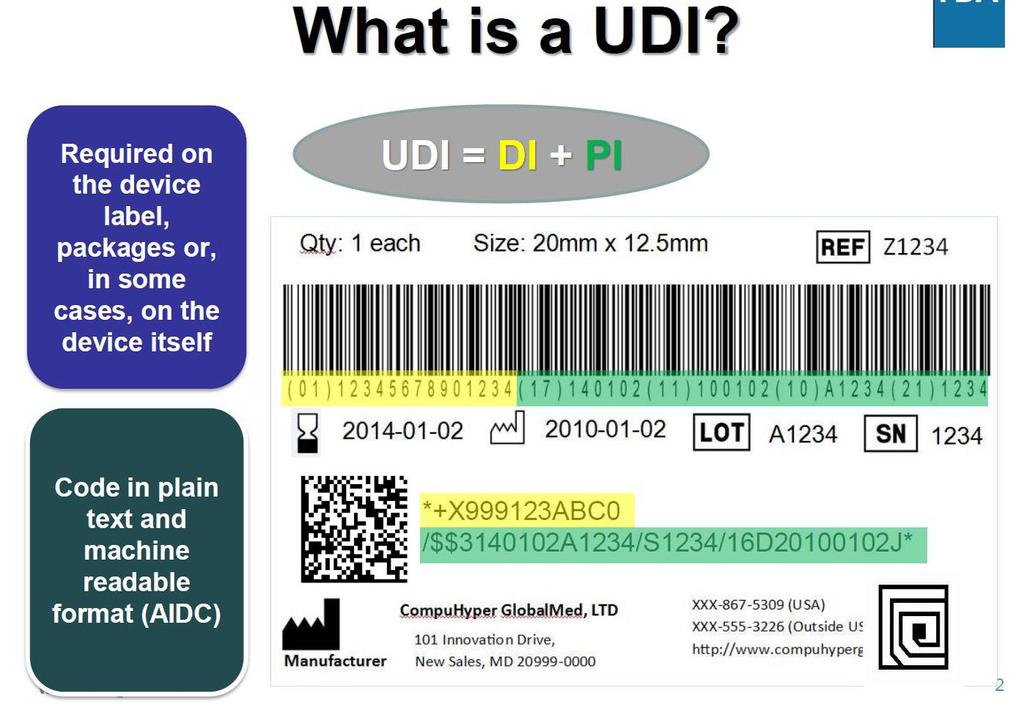
46

47 Produsenter-importører-distributører og skal lagre UDI-koden. Hva med helsetjenesten?
48 Sporbarhet i medisinsk utstyrs kjeden Produsent importør Bruker distributør I dag: Håndterings Forskriften meldeplikten uhell. «identititeten til det medisinsk utstyret skal kunne spores». Fremtiden: Helseinstitusjoner skal lagre og oppbevare UDI`en for implanterbart medisinsk utstyr klasse III.
49 Helseinstitusjoner Nytt regelverk gir at helseinstitusjoner skal, helst i elektronisk form, lagre og oppbevare UDI'en for implanterbart medisinsk utstyr i klasse III som de har utlevert eller som de har mottatt. For annet utstyr enn implanterbart medisinsk utstyr i klasse III kan medlemsstatene enten oppfordre til eller kreve at helseinstitusjoner lagrer og oppbevarer UDI'en. I tillegg kan medlemsstatene både oppfordre til og kreve at helsepersonell skal lagre og oppbevare UDI'en for utstyr som de har mottatt
50 Implantatkort Produsenten skal utlevere et implantatkort sammen med implanterbart medisinsk utstyret Kortet identifiserer utstyret og inneholder nødvendige advarsler/forholdsregler, herunder utstyrets forventede levetid. Følgende implantater er unntatt (dansk forordningstekst): hæfteklammer, tandfyldninger, tandbøjler, tandkroner, skruer, kiler, plader, tråd, nåle, clips og forbindelsesled. Kommisjonen kan endre listen over implantater med gjennomføringsrettsakter. Medisinsk utstyr nye forordninger 50
51 Operators UDI Clinical evaluation Products Notified Body EUDAMED Vigilance Certificate Clinical investigations Market surveillance
52 EUDAMED/UDI Ett nøkkelaspekt for å oppfylle formålene med forordningene er videreutvikling og forvaltning av den felles europeiske databasen for medisinsk utstyr (EUDAMED) Kommisjonen skal videreutvikle og forvalte den europeiske databasen for medisinsk utstyr som ble opprettet ved Kommisjonsvedtak 2010/227/EU. Kommisjonen skal ved design av EUDAMED ta hensyn til kompatibilitet med nasjonale databaser og nasjonal webgrensesnitt for å kunne importere og eksportere relevante data. Dataene skal registreres av medlemsstatene, tekniske kontrollorgan, markedsaktører og sponsorer
53 EUDAMED Kommisjonen og MDCG (Medical Device Coordination Group) har utarbeidet en plan for gjennomføring av funksjonsspesifikasjonene. Planen har som formål å sikre at EUDAMED er fullt ut operativ innen 25. mars 2020.
54 EUDAMED består av ulike databaser Registrering av utstyr (MDR artikkel 29 stk. 4/IVDR art. 26) UDI-databasen (MDR art. 28/IVDR art. 26) Registrering av markedsdeltakere (MDR art. 30/IVDR art. 27) Tekniske kontrollorgan og sertifikater (MDR art. 57/IVDR art. 52) Kliniske utprøvinger/vurdering av utstyrets ytelse (MDR art. 73/IVDR art. 69) Markedsovervåking av utstyr etter brakt i omsetning (MDR art. 92/IVDR art. 87) Markedsovervåking (MDR art. 100/IVDR art. 95)
55 Markedsovervåking av utstyr etter brakt i omsetning Som ledd i kvalitetssystemet skal produsenten skal utarbeide en plan for overvåking etter at utstyret er plassert på markedet. Planen skal inneholde opplysningene i MDR/IVDR vedlegg III, punkt 1 og skal være en del av den tekniske dokumentasjonen jf. MDR/IVDR vedlegg II. Planen skal blant annet inneholde opplysninger om alvorlige hendelser, et register over ikke-alvorlige hendelser og data om uønskede bivirkninger, melding om trender, klager fra brukere, distributører og importører mv. Planen skal beskrive en proaktiv og systematisk prosess for innsamling av opplysningene og inneholde en metodikk for å vurdere dataene og evt. iverksette korrigerende tiltak ved behov MDR art. 84/IVDR art. 79
56 Markedsovervåking av utstyr etter brakt i omsetning Produsenter av medisinsk utstyr i klasse I (MDR) og klasse A og B (IVDR) skal utarbeide en rapport om overvåking etter at utstyr er plassert på markedet. Rapporten (Post market surveillance report) skal oppsummere resultatene og konklusjonene av funnene i planen nevnt ovenfor. Rapporten skal oppdateres etter behov og på anmodning stilles til rådighet for kompetente myndigheter. Produsenter av utstyr i klasse IIa, IIb og III (MDR) og klasse C og D (IVDR) skal utarbeide en periodisk oppdatert sikkerhetsrapport (Periodic safety update report, forkortet PSUR) for hvert utstyr/hver kategori eller gruppe av utstyr. PSUR skal inneholde et sammendrag av resultatene og konklusjonene fra overvåking etter at utstyr er plassert på markedet.
57 Vigilance (meldesystemet) Prinsippet om at produsenten er juridisk ansvarlig for å melde om hendelser videreføres i MDR/IVDR. Produsenter skal melde om følgende hendelser med det medisinske utstyret til kompetente myndigheter (unntatt utstyr til klinisk utprøving/utstyr til vurdering av ytelse): a) enhver alvorlig hendelse med utstyr som er omsatt på EU/EØSmarkedet, unntatt forventede bivirkninger som klart er dokumentert i produktinformasjonen og beskrevet i teknisk dokumentasjon. b) ethvert sikkerhetsrelatert korrigerende tiltak (field safety corrective action) med utstyr som er plassert på EU/EØS-markedet. Meldingene skal sendes inn via EUDAMED
58 Trender Produsenter skal via EUDAMED melde om enhver statistisk signifikant økning i frekvensen av hendelser, som ikke er alvorlige hendelser eller forventede uønskede bivirkninger, som kan ha vesentlig betydning for vurdering av forholdet mellom fordeler og risiko i MDR/IVDR vedlegg I punkt 1 og 5 og som har eller som kan ha medført risiko for pasienter, brukere elle andre personers helse og sikkerhet
59 Key dates 26. november 2017: Kravene om tekniske kontrollorgan, utpeking av kompetente myndigheter og etablering av MDCG skal være oppfylt 26. mai 2018: Kravene om samarbeid mellom myndighetene etter forordningen skal være påbegynt 26. november 2023 : Registrering av utstyr etter IVDR gjelder 26. mai 2023 : Kravene til sporbarhet og UDI trer i kraft 26. mai 2024: Gyldigheten til sertifikater etter gjeldende EU-direktiver utløper 26. mai 2025: Siste frist for å omsette medisinsk utstyr på markedet etter gjeldende EU-direktiver 26. mai 2027: Koordinerende prosedyre for kliniske utprøvinger starter. Legemiddelverket

60 Hva gjøres av norske myndigheter Medisinsk utstyr nye forordninger 60
61 Medisinsk utstyr nye forordninger 61
62 Hvordan jobber norske myndigheter? De nye forordningene vil bli tatt inn i EØS-avtalen Regelverket vil kreve endring i lov og forskrift om medisinsk utstyr og i nasjonal forskrift om håndtering av medisinsk utstyr. Legemiddelverket har utarbeidet høringsnotat for gjennomføring av forordningene i norsk rett. Høringsnotatet vil deretter bli godkjent av Helse- og omsorgsdepartementet og sendes på offentlig høring. HOD tar sikte på ikrafttredelse i Norge samtidig som i EU 26. mai Enhet for EØS-oversettelse i Utenriksdepartementet arbeider med å oversette forordningene til norsk 62
63 Hva blir de største utfordringene i forbindelse med implementering frem til 2020?
64 Operators UDI Clinical evaluation Market surveillance Products Notified Body Vigilance MDR/IVDR JA/JAMS Governance Clinical investigations MDD/ IVDD
65 26. mai MDR 19 kalender måneder fra nå Kommisjonen jobber med å få på plass: - Eudamed - Harmoniserte standarder - Common Specifications - Implementing Acts - Ekspert Paneler - EU Referanse labortaorier - Tekniske kontrollorgan 26. mai IVDR 43 kalender måneder fra nå
66 Utfordringer Utstyr som må gjennom teknisk kontrollorgan for første gang (ca. 85%) IVD Eksempler: Fertility, pregnancy and sexually-transmitted disease tests Klasse III implanterbart individuelt tilpasset medisinsk utstyr Eksempler: implants (hip liners, knee inserts, etc.) Gjenbrukbare kirurgiske instrumenter som før var klasse I utstyr (samsvarserklæring) Example: Reusable scalpels for routine surgery Medisinsk utstyr som inneholder nanomateriale som før var klasse I (samsvarserklæring) Software MD som før var klasse I utstyr I self-certified Eksempel: Dental imaging software
67 For produsenter Forstå MDR/IVDR Hva med QMS systemet ditt Gyldigheten av klinisk dokumentasjon Betydning for ditt produkt Endring teknisk dokumentasjon Produktmerking Sjekk MUdefinisjon Sjekk klassifisering Post market surveillance
68 For produsenter Post market clinical follow up plan Hva med dine distributører/i mportører Når utløper sertifikatetet? Hva med vigilance. Hendelser? Økonomisk dekning for skader/erstat ning Kontakt ditt tekniske kontrollorgan Følg med på lovendringer Sporbarhet av utstyret Ansvarlig person for «compliance» Opplæring ansatte
69 Hvor finnes relevant veiledningsmateriell fra EU Factsheet for manufacturers of medical devices Implementation model: medical devices Exhaustive list: requirements for medical devices manufacturers Implementation model: in-vitro diagnostic medical devices MDCG documents New regulations CAMD Transitional: CAMD Implemetation Taskforce MDR/IVDR Roadmap

70 Takk for oss! Lese mer? Spørsmål? Medisinsk utstyr nye forordninger 70




71 Følg legemiddelverket legemiddelverket.no
Nye forordninger om medisinsk utstyr (MDR-IVDR) Hva så?
 Hva så?") Nye forordninger om medisinsk utstyr (MDR-IVDR) Hva så? Petter Alexander Strømme Bjørn Kristian Berge 26. mai 2020 Ny forordning om medisinsk utstyr 26. mai 2022 Ny forordning om invitro diagnostisk medisinsk
Nye forordninger om medisinsk utstyr (MDR-IVDR) Hva så? Petter Alexander Strømme Bjørn Kristian Berge 26. mai 2020 Ny forordning om medisinsk utstyr 26. mai 2022 Ny forordning om invitro diagnostisk medisinsk
Ny forordning om in vitro diagnostisk medisinsk utstyr
 Ny forordning om in vitro diagnostisk medisinsk utstyr Petter Alexander Strømme & Bjørn Kristian Berge avdeling medisinsk utstyr & legemidler Akkrediteringsdag for medisinske laboratorier i Lillestrøm
Ny forordning om in vitro diagnostisk medisinsk utstyr Petter Alexander Strømme & Bjørn Kristian Berge avdeling medisinsk utstyr & legemidler Akkrediteringsdag for medisinske laboratorier i Lillestrøm
regelverket for medisinsk utstyr og legemiddelverkets rolle som fagmyndighet.
 informasjonsmøte 8. mai 2018 regelverket for medisinsk utstyr og legemiddelverkets rolle som fagmyndighet. Ingeborg Hagerup-Jenssen, Statens legemiddelverk litt om. vår rolle hva er medisinsk utstyr dagens
informasjonsmøte 8. mai 2018 regelverket for medisinsk utstyr og legemiddelverkets rolle som fagmyndighet. Ingeborg Hagerup-Jenssen, Statens legemiddelverk litt om. vår rolle hva er medisinsk utstyr dagens
QA Regulatory Forum 5. april medical devices - medisinsk utstyr MDR & IVDR, classification ++ government perspective
 medical devices - medisinsk utstyr MDR & IVDR, classification ++ government perspective Petter Alexander Strømme & Ingeborg Hagerup-Jenssen, Statens legemiddelverk litt om. hvem vi er & hva vi gjør MDR
medical devices - medisinsk utstyr MDR & IVDR, classification ++ government perspective Petter Alexander Strømme & Ingeborg Hagerup-Jenssen, Statens legemiddelverk litt om. hvem vi er & hva vi gjør MDR
Høringsmøte om forslag til nye regler om medisinsk utstyr
 Høringsmøte om forslag til nye regler om medisinsk utstyr Program 12:30 13:00 Velkommen og innledning fra Helse - og omsorgsdepartementet 13:00 13:40 Presentasjon av høringsforslag 13:40 14:00 Pause og
Høringsmøte om forslag til nye regler om medisinsk utstyr Program 12:30 13:00 Velkommen og innledning fra Helse - og omsorgsdepartementet 13:00 13:40 Presentasjon av høringsforslag 13:40 14:00 Pause og
Er sykehus og sykehjem klare for nytt regelverk? Petter Alexander Strømme og Bjørn Kristian Berge seniorrådgivere medisinsk utstyr, Legemiddelverket
 Er sykehus og sykehjem klare for nytt regelverk? Petter Alexander Strømme og Bjørn Kristian Berge seniorrådgivere medisinsk utstyr, Legemiddelverket Nasjonal fagmyndighet for medisinsk utstyr Forvaltnings-
Er sykehus og sykehjem klare for nytt regelverk? Petter Alexander Strømme og Bjørn Kristian Berge seniorrådgivere medisinsk utstyr, Legemiddelverket Nasjonal fagmyndighet for medisinsk utstyr Forvaltnings-
Medisinsk utstyr. Katrine S. Edvardsen Espantaleón og Bjørn Kristian Berge Avd. medisinsk utstyr og legemidler
 Medisinsk utstyr Katrine S. Edvardsen Espantaleón og Bjørn Kristian Berge Avd. medisinsk utstyr og legemidler Nasjonal fagmyndighet for medisinsk utstyr Forvaltnings- og rådgivningsoppgaver Tilsynsmyndighet
Medisinsk utstyr Katrine S. Edvardsen Espantaleón og Bjørn Kristian Berge Avd. medisinsk utstyr og legemidler Nasjonal fagmyndighet for medisinsk utstyr Forvaltnings- og rådgivningsoppgaver Tilsynsmyndighet
Høring om gjennomføring av forordning 2017/745 om medisinsk utstyr og forordning 2017/746 om in vitro-diagnostisk medisinsk utstyr
 Høring om gjennomføring av forordning 2017/745 om medisinsk utstyr og forordning 2017/746 om in vitro-diagnostisk medisinsk utstyr Ny lov og forskrift om medisinsk utstyr Høringsfrist: 23. august 2019
Høring om gjennomføring av forordning 2017/745 om medisinsk utstyr og forordning 2017/746 om in vitro-diagnostisk medisinsk utstyr Ny lov og forskrift om medisinsk utstyr Høringsfrist: 23. august 2019
Håndteringsforskriften i praksis for gjenbruksinstrumenter. Avdeling for medisinsk utstyr og legemidler
 Håndteringsforskriften i praksis for gjenbruksinstrumenter Avdeling for medisinsk utstyr og legemidler 2 Nasjonal fagmyndighet for medisinsk utstyr Forvaltnings- og rådgivningsoppgaver Tilsynsmyndighet
Håndteringsforskriften i praksis for gjenbruksinstrumenter Avdeling for medisinsk utstyr og legemidler 2 Nasjonal fagmyndighet for medisinsk utstyr Forvaltnings- og rådgivningsoppgaver Tilsynsmyndighet
In-house medisinsk utstyr. Medisinsk utstyr som produseres og brukes internt i helseinstitusjon
 In-house medisinsk utstyr Medisinsk utstyr som produseres og brukes internt i helseinstitusjon CE-merket medisinsk utstyr Produsent Produktutvikling og produksjon Samsvar med krav til ytelse og sikkerhet
In-house medisinsk utstyr Medisinsk utstyr som produseres og brukes internt i helseinstitusjon CE-merket medisinsk utstyr Produsent Produktutvikling og produksjon Samsvar med krav til ytelse og sikkerhet
Hva er MDR. Hvordan er den knyttet til GS1 standard, e-helse og IKT i helsesektoren?
 Hva er MDR Hvordan er den knyttet til GS1 standard, e-helse og IKT i helsesektoren? Nasjonal fagmyndighet for medisinsk utstyr Forvaltnings- og rådgivningsoppgaver Tilsynsmyndighet overfor produsenter,
Hva er MDR Hvordan er den knyttet til GS1 standard, e-helse og IKT i helsesektoren? Nasjonal fagmyndighet for medisinsk utstyr Forvaltnings- og rådgivningsoppgaver Tilsynsmyndighet overfor produsenter,
Innhold. Helsedirektoratet. Etablert 1. januar 2002 Organ for iverksetting av nasjonal politikk på helseområdet
 Regelverket for in vitro diagnostisk (IVD) medisinsk utstyr Bjørn Kristian Berge Innhold Kort om Helsedirektoratet Regelverket for in vitro diagnostisk (IVD) medisinsk utstyr Helsedirektoratets rolle Definisjoner
Regelverket for in vitro diagnostisk (IVD) medisinsk utstyr Bjørn Kristian Berge Innhold Kort om Helsedirektoratet Regelverket for in vitro diagnostisk (IVD) medisinsk utstyr Helsedirektoratets rolle Definisjoner
Endringer i forskrift 20. desember 2007 nr om målenheter og måling. Implementering av nye MID- og NAWI-direktiver.
 Endringer i forskrift 20. desember 2007 nr. 1723 om målenheter og måling. Implementering av nye MID- og NAWI-direktiver. 1. Hovedinnholdet i forslaget Forslag til endringer i forskrift 20. desember 2007
Endringer i forskrift 20. desember 2007 nr. 1723 om målenheter og måling. Implementering av nye MID- og NAWI-direktiver. 1. Hovedinnholdet i forslaget Forslag til endringer i forskrift 20. desember 2007
Regelverket for medisinsk utstyr status og kommende endringer. seniorrådgiver Ingeborg Hagerup-Jenssen avdeling medisinsk utstyr og legemidler
 Regelverket for medisinsk utstyr status og kommende endringer seniorrådgiver Ingeborg Hagerup-Jenssen avdeling medisinsk utstyr og legemidler Regelverket i dag Nært forestående endringer og mulige implikasjoner
Regelverket for medisinsk utstyr status og kommende endringer seniorrådgiver Ingeborg Hagerup-Jenssen avdeling medisinsk utstyr og legemidler Regelverket i dag Nært forestående endringer og mulige implikasjoner
Nasjonal fagmyndighet for medisinsk utstyr
 Produksjon av IVD medisinsk utstyr i laboratoriet Eli Neegaard og Bjørn Kristian Berge Avdeling for medisinsk utstyr og legemidler Akkrediteringsdagen 02.12.2015 Nasjonal fagmyndighet for medisinsk utstyr
Produksjon av IVD medisinsk utstyr i laboratoriet Eli Neegaard og Bjørn Kristian Berge Avdeling for medisinsk utstyr og legemidler Akkrediteringsdagen 02.12.2015 Nasjonal fagmyndighet for medisinsk utstyr
Introduksjon av nye produkter hva er viktig å være oppmerksom på?
 Kvalitetssikring av innkjøpsprosesser i Helse Sør-Øst 28. Januar 2015 Introduksjon av nye produkter hva er viktig å være oppmerksom på? Avdeling for smittevern Disposisjon Før bestiling/anbud Behovsanalyse
Kvalitetssikring av innkjøpsprosesser i Helse Sør-Øst 28. Januar 2015 Introduksjon av nye produkter hva er viktig å være oppmerksom på? Avdeling for smittevern Disposisjon Før bestiling/anbud Behovsanalyse
Melding om høring - endring i forskrift om medisinsk utstyr
 v4-29.07.2015 Returadresse: Helsedirektoratet, Pb. 7000 St. Olavs plass, 0130 Oslo, Norge HDIR Innland 25125808 Høringsinstanser Deres ref.: Vår ref.: 17/29839-3 Saksbehandler: Petter Alexander Strømme
v4-29.07.2015 Returadresse: Helsedirektoratet, Pb. 7000 St. Olavs plass, 0130 Oslo, Norge HDIR Innland 25125808 Høringsinstanser Deres ref.: Vår ref.: 17/29839-3 Saksbehandler: Petter Alexander Strømme
Til 1-2 (forskriftens virkeområde) Til første ledd. Der hvor det i forskriften vises til utstyr menes både medisinsk utstyr og tilbehør.
 Til første ledd. Der hvor det i forskriften vises til utstyr menes både medisinsk utstyr og tilbehør.") Merknader til enkelte av bestemmelsene i forskrift om medisinsk utstyr Merknadene er en veiledning for å utdype innholdet i den enkelte bestemmelse i forskriften. Merknadene er i seg selv ikke rettslig
Merknader til enkelte av bestemmelsene i forskrift om medisinsk utstyr Merknadene er en veiledning for å utdype innholdet i den enkelte bestemmelse i forskriften. Merknadene er i seg selv ikke rettslig
Klikk ikonet for å legge til et bilde. Medisinsk utstyr. Regelverk og roller. Landsmøte Norsk forening for Sterilforsyning 5.
 Klikk ikonet for å legge til et bilde Medisinsk utstyr Regelverk og roller for Sterilforsyning 5. juni 2015, Nasjonal fagmyndighet for medisinsk utstyr Forvaltnings- og rådgivningsoppgaver Tilsynsmyndighet
Klikk ikonet for å legge til et bilde Medisinsk utstyr Regelverk og roller for Sterilforsyning 5. juni 2015, Nasjonal fagmyndighet for medisinsk utstyr Forvaltnings- og rådgivningsoppgaver Tilsynsmyndighet
Utkast til forskrift om håndtering av medisinsk utstyr
 Utkast til forskrift om håndtering av medisinsk utstyr Hjemmel: Fastsatt ved kgl.res xx.xx.201x med hjemmel i lov 24. mai 1929 nr. 4 om tilsyn med elektriske anlegg og elektrisk utstyr 10, lov 2. juli
Utkast til forskrift om håndtering av medisinsk utstyr Hjemmel: Fastsatt ved kgl.res xx.xx.201x med hjemmel i lov 24. mai 1929 nr. 4 om tilsyn med elektriske anlegg og elektrisk utstyr 10, lov 2. juli
NOR/306R T OJ X 92/06, p. 6-9
 NOR/306R0507.00T OJ X 92/06, p. 6-9 COMMISSION REGULATION (EC) No 507/2006 of 29 March 2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of Regulation
NOR/306R0507.00T OJ X 92/06, p. 6-9 COMMISSION REGULATION (EC) No 507/2006 of 29 March 2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of Regulation
II. Krav til byggevarer som er CE-merket
 II. Krav til byggevarer som er CE-merket Lastet ned fra Direktoratet for byggkvalitet 27.10.2015 II. Krav til byggevarer som er CE-merket Innledning Kapittel II gjennomfører byggevareforordningen (forordning
II. Krav til byggevarer som er CE-merket Lastet ned fra Direktoratet for byggkvalitet 27.10.2015 II. Krav til byggevarer som er CE-merket Innledning Kapittel II gjennomfører byggevareforordningen (forordning
CE-merking EU «In Vitro Diagnostikk-direktivet» v/kari Velsand, 15. mars 2018
 CE-merking EU «In Vitro Diagnostikk-direktivet» v/kari Velsand, 15. mars 2018 Holder for related picture Agenda Introduksjon Lovgivning for medisinsk utstyr i EU Krav i IVD-direktivet 2 Introduksjon Hva
CE-merking EU «In Vitro Diagnostikk-direktivet» v/kari Velsand, 15. mars 2018 Holder for related picture Agenda Introduksjon Lovgivning for medisinsk utstyr i EU Krav i IVD-direktivet 2 Introduksjon Hva
R2007.hza
 Nedenfor gjengis til informasjon EØS-avtalen vedlegg I kapittel I del 7.2 nr. 41 (forordning (EF) nr. 2007/2006) slik Mattilsynet tolker denne del av EØS-avtalen med de endringer og tillegg som følger
Nedenfor gjengis til informasjon EØS-avtalen vedlegg I kapittel I del 7.2 nr. 41 (forordning (EF) nr. 2007/2006) slik Mattilsynet tolker denne del av EØS-avtalen med de endringer og tillegg som følger
Byggevareforordningens krav 0l CE- merking og dokumentasjonskravet 0l ikke CE- merkede. HANNE PRESTMO 06.11.13 RørArena
 Byggevareforordningens krav 0l CE- merking og dokumentasjonskravet 0l ikke CE- merkede produkter HANNE PRESTMO 06.11.13 RørArena Presentasjonens innhold 1. Regelverket i dag 2. Skillet mellom omsetning
Byggevareforordningens krav 0l CE- merking og dokumentasjonskravet 0l ikke CE- merkede produkter HANNE PRESTMO 06.11.13 RørArena Presentasjonens innhold 1. Regelverket i dag 2. Skillet mellom omsetning
HØRINGSNOTAT OM forlag TIL ENDRINGER I DOK- FORSKRIFTEN
 HØRINGSNOTAT OM forlag TIL ENDRINGER I DOKFORSKRIFTEN Ved alminnelig høring av utkast til forskrift om endring til forskrift om dokumentasjon og omsetning av produkter til byggverk (DOKforskriften) av
HØRINGSNOTAT OM forlag TIL ENDRINGER I DOKFORSKRIFTEN Ved alminnelig høring av utkast til forskrift om endring til forskrift om dokumentasjon og omsetning av produkter til byggverk (DOKforskriften) av
REGULATION (EU) 2017/745 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation
 2017/745 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation") REGULATION (EU) 2017/745 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and
REGULATION (EU) 2017/745 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and
Leketøyseminar 2013. Nytt og viktig!
 Leketøyseminar 2013 Nytt og viktig! Berit Jaritz, DSB 1 Leketøyforskriften = fellesforskrift To myndigheter å forholde seg til: DSB: Fysiske, mekaniske, brann- og eksplosjon, elektriske og radioaktive
Leketøyseminar 2013 Nytt og viktig! Berit Jaritz, DSB 1 Leketøyforskriften = fellesforskrift To myndigheter å forholde seg til: DSB: Fysiske, mekaniske, brann- og eksplosjon, elektriske og radioaktive
AROMATERAPI OG REGELVERK
 AROMATERAPI OG REGELVERK Kjemikalier er en felles betegnelse for kjemiske stoffer og stoffblandinger, som både forekommer i naturlig tilstand og som framstilles industrielt. Reglene for klassifisering,
AROMATERAPI OG REGELVERK Kjemikalier er en felles betegnelse for kjemiske stoffer og stoffblandinger, som både forekommer i naturlig tilstand og som framstilles industrielt. Reglene for klassifisering,
Høring Europakommisjonens forslag til forordning om gjensidig godkjenning av varer lovlig omsatt i et annet medlemsland
 Høringsnotat Dato: 6. februar 2018 Saksnr.: 18/702 Høring Europakommisjonens forslag til forordning om gjensidig godkjenning av varer lovlig omsatt i et annet medlemsland 1. Innledning Nærings- og fiskeridepartementet
Høringsnotat Dato: 6. februar 2018 Saksnr.: 18/702 Høring Europakommisjonens forslag til forordning om gjensidig godkjenning av varer lovlig omsatt i et annet medlemsland 1. Innledning Nærings- og fiskeridepartementet
Velkommen. Maskindirektivet. Når kompetanse teller.. Innhold
 Velkommen Maskindirektivet Lillestrøm 10. nov. 2016 Foredragsholder: Navn: Jørn Erik Andersen Mob. 95 08 30 83 Når kompetanse teller.. Innhold Direktiv forskrift og norm Forskjell på elektrisk anlegg og
Velkommen Maskindirektivet Lillestrøm 10. nov. 2016 Foredragsholder: Navn: Jørn Erik Andersen Mob. 95 08 30 83 Når kompetanse teller.. Innhold Direktiv forskrift og norm Forskjell på elektrisk anlegg og
Forslag til ny lov om behandling av personopplysninger
 Justis- og beredskapsdepartementet Forslag til ny lov om behandling av personopplysninger Personvernkonferansen 8. desember 2017 Anne Sofie Hippe, fung. Lovrådgiver, Oversikt Kort om personvernforordningen
Justis- og beredskapsdepartementet Forslag til ny lov om behandling av personopplysninger Personvernkonferansen 8. desember 2017 Anne Sofie Hippe, fung. Lovrådgiver, Oversikt Kort om personvernforordningen
Akkreditering sertifisering
 Akkreditering sertifisering Tromsø, 11. 12. februar 2015 Kari Iversen Dyrdal Kvalitetsleder Avdeling for immunologi og transfusjonsmedisin Sertifisering ISO 9001:2008 (kvalitetsledelse) Akkreditering ISO
Akkreditering sertifisering Tromsø, 11. 12. februar 2015 Kari Iversen Dyrdal Kvalitetsleder Avdeling for immunologi og transfusjonsmedisin Sertifisering ISO 9001:2008 (kvalitetsledelse) Akkreditering ISO
Forskrift om dokumentasjon av byggevarer Definisjoner
 Definisjoner Lastet ned fra Direktoratet for byggkvalitet 28.08.2015 Definisjoner Lastet ned fra Direktoratet for byggkvalitet 28.08.2015 2 Definisjon Autorisert representant Byggevare Byggesett Byggevareinfo.no
Definisjoner Lastet ned fra Direktoratet for byggkvalitet 28.08.2015 Definisjoner Lastet ned fra Direktoratet for byggkvalitet 28.08.2015 2 Definisjon Autorisert representant Byggevare Byggesett Byggevareinfo.no
Bygg produktforskriften
 Bygg produktforskriften Obligatorisk 1. juli 2013 -merking CE-merking er et pass som gjør at et produkt kan plasseres lovlig på markedet i noen medlemsland. Fra 1. juli 2013 starter CE-merkingen som skal
Bygg produktforskriften Obligatorisk 1. juli 2013 -merking CE-merking er et pass som gjør at et produkt kan plasseres lovlig på markedet i noen medlemsland. Fra 1. juli 2013 starter CE-merkingen som skal
Gjennomføring av europaparlaments- og rådsforordning (EU) nr. 536/2014 om klinisk utprøving av legemidler til mennesker i norsk rett.
 nr. 536/2014 om klinisk utprøving av legemidler til mennesker i norsk rett.") 1 Høringsnotat Gjennomføring av europaparlaments- og rådsforordning (EU) nr. 536/2014 om klinisk utprøving av legemidler til mennesker i norsk rett. 1 Innledning. EØS-komiteen vedtok ved beslutning nr.
1 Høringsnotat Gjennomføring av europaparlaments- og rådsforordning (EU) nr. 536/2014 om klinisk utprøving av legemidler til mennesker i norsk rett. 1 Innledning. EØS-komiteen vedtok ved beslutning nr.
Ny Plan- og bygningslov med tilhørende forskrifter. Else Øvernes, KRD
 Ny Plan- og bygningslov med tilhørende forskrifter Else Øvernes, KRD 1 Tema Krav om uavhengig kontroll og tilsyn i byggesaker Krav til produktdokumentasjon i ny teknisk forskrift Markedstilsyn og myndighetenes
Ny Plan- og bygningslov med tilhørende forskrifter Else Øvernes, KRD 1 Tema Krav om uavhengig kontroll og tilsyn i byggesaker Krav til produktdokumentasjon i ny teknisk forskrift Markedstilsyn og myndighetenes
NOR/31R0143.ohfo OJ L 44/11, p. 2-6
 NOR/31R0143.ohfo OJ L 44/11, p. 2-6 COMMISSION REGULATION (EU) No 143/2011 of 17 February 2011 amending Annex XIV to Regulation (EC) No 1907/2006 of the European Parliament and of the Council on the Registration,
NOR/31R0143.ohfo OJ L 44/11, p. 2-6 COMMISSION REGULATION (EU) No 143/2011 of 17 February 2011 amending Annex XIV to Regulation (EC) No 1907/2006 of the European Parliament and of the Council on the Registration,
(UOFFISIELL OVERSETTELSE)
") NOR/312R0207.tona OJ L 72/2012, p. 28-31 COMMISSION REGULATION (EU) No 207/2012 of 9 March 2012 on electronic instructions for use of medical devices. (UOFFISIELL OVERSETTELSE) EUROPAKOMMISJONEN HAR KOMMISJONSFORORDNING
NOR/312R0207.tona OJ L 72/2012, p. 28-31 COMMISSION REGULATION (EU) No 207/2012 of 9 March 2012 on electronic instructions for use of medical devices. (UOFFISIELL OVERSETTELSE) EUROPAKOMMISJONEN HAR KOMMISJONSFORORDNING
Forskrift om EØS-krav til sikkerhet for utstyr til elektronisk kommunikasjon
 Forskrift om EØS-krav til sikkerhet for utstyr til elektronisk kommunikasjon Forskriftens hjemmels- og EØS-henvisningsfelt skal lyde: Hjemmel: Fastsatt av Nasjonal kommunikasjonsmyndighet dd.mm.2016 med
Forskrift om EØS-krav til sikkerhet for utstyr til elektronisk kommunikasjon Forskriftens hjemmels- og EØS-henvisningsfelt skal lyde: Hjemmel: Fastsatt av Nasjonal kommunikasjonsmyndighet dd.mm.2016 med
Høringsnotat. Forslag til supplerende bestemmelser om sikkerhetsanordninger
 Høringsnotat Forslag til supplerende bestemmelser om sikkerhetsanordninger Legemiddelverket sender med dette på vegne av Helse- og omsorgsdepartementet på høring forslag til ytterligere bestemmelser for
Høringsnotat Forslag til supplerende bestemmelser om sikkerhetsanordninger Legemiddelverket sender med dette på vegne av Helse- og omsorgsdepartementet på høring forslag til ytterligere bestemmelser for
NORSK LOVTIDEND Avd. I Lover og sentrale forskrifter mv. Utgitt i henhold til lov 19. juni 1969 nr. 53.
 NORSK LOVTIDEND Avd. I Lover og sentrale forskrifter mv. Utgitt i henhold til lov 19. juni 1969 nr. 53. Kunngjort 24. august 2018 kl. 13.35 PDF-versjon 29. august 2018 21.08.2018 nr. 1262 Forskrift om
NORSK LOVTIDEND Avd. I Lover og sentrale forskrifter mv. Utgitt i henhold til lov 19. juni 1969 nr. 53. Kunngjort 24. august 2018 kl. 13.35 PDF-versjon 29. august 2018 21.08.2018 nr. 1262 Forskrift om
Høring Gjennomføring av forordning (EU) 2016/426 om gassapparater og om opphevelse av direktiv 2009/142/EF
 2016/426 om gassapparater og om opphevelse av direktiv 2009/142/EF") Høringsnotat Høring Gjennomføring av forordning (EU) 2016/426 om gassapparater og om opphevelse av direktiv 2009/142/EF 1. Bakgrunnen for forslaget Europaparlamentet og Rådet vedtok 9. mars 2016 forordning
Høringsnotat Høring Gjennomføring av forordning (EU) 2016/426 om gassapparater og om opphevelse av direktiv 2009/142/EF 1. Bakgrunnen for forslaget Europaparlamentet og Rådet vedtok 9. mars 2016 forordning
COMMISSION REGULATION (EU) No 488/2012 of 8 June 2012 amending Regulation (EC) No 658/2007 concerning financial penalties for infringement of certain
 No 488/2012 of 8 June 2012 amending Regulation (EC) No 658/2007 concerning financial penalties for infringement of certain") COMMISSION REGULATION (EU) No 488/2012 of 8 June 2012 amending Regulation (EC) No 658/2007 concerning financial penalties for infringement of certain obligations in connection with marketing authorisations
COMMISSION REGULATION (EU) No 488/2012 of 8 June 2012 amending Regulation (EC) No 658/2007 concerning financial penalties for infringement of certain obligations in connection with marketing authorisations
(UOFFISIELL OVERSETTELSE)
") NOR/312R1237.fral COMMISSION IMPLEMENTING REGULATION (EU) No 1237/2012 of 19 December 2012 approving the active substance Zucchini Yellow Mosaic Virus weak strain, in accordance with Regulation (EC) No
NOR/312R1237.fral COMMISSION IMPLEMENTING REGULATION (EU) No 1237/2012 of 19 December 2012 approving the active substance Zucchini Yellow Mosaic Virus weak strain, in accordance with Regulation (EC) No
Biocider. Biocidforskriften og desinfeksjonsmidler i helsevesenet
 Biocider Biocidforskriften og desinfeksjonsmidler i helsevesenet Jorid Frydenlund Seksjon for biocider og globalt kjemikaliearbeid Miljøgiftavdelingen Tema Hva er et biocid Regelverk Markedsføring av biocidprodukter
Biocider Biocidforskriften og desinfeksjonsmidler i helsevesenet Jorid Frydenlund Seksjon for biocider og globalt kjemikaliearbeid Miljøgiftavdelingen Tema Hva er et biocid Regelverk Markedsføring av biocidprodukter
(UOFFISIELL OVERSETTELSE)
") NOR/313R0305.eltr OJ L 91/13, p. 1-4 COMMISSION DELEGATED REGULATION (EU) No 305/2013 of 26 November 2012 supplementing Directive 2010/40/EU of the European Parliament and of the Council with regard to
NOR/313R0305.eltr OJ L 91/13, p. 1-4 COMMISSION DELEGATED REGULATION (EU) No 305/2013 of 26 November 2012 supplementing Directive 2010/40/EU of the European Parliament and of the Council with regard to
Common Safety Methods
 Common Safety Methods Johan L. Aase Sikkerhets- og Kvalitetssjef Utbyggingsdivisjonen Jernbaneverket ESRA - 11.11.09 Foto: RuneFossum,Jernbanefoto.no CSM Common Safety methods Common Safety Method on Risk
Common Safety Methods Johan L. Aase Sikkerhets- og Kvalitetssjef Utbyggingsdivisjonen Jernbaneverket ESRA - 11.11.09 Foto: RuneFossum,Jernbanefoto.no CSM Common Safety methods Common Safety Method on Risk
Nr. 49/254 EØS-tillegget til Den europeiske unions tidende KOMMISJONSDIREKTIV 2008/43/EF. av 4. april 2008
 Nr. 49/254 EØS-tillegget til Den europeiske unions tidende KOMMISJONSDIREKTIV 2008/43/EF 2015/EØS/49/49 av 4. april 2008 om opprettelse av et system for identifikasjon og sporing av eksplosive varer til
Nr. 49/254 EØS-tillegget til Den europeiske unions tidende KOMMISJONSDIREKTIV 2008/43/EF 2015/EØS/49/49 av 4. april 2008 om opprettelse av et system for identifikasjon og sporing av eksplosive varer til
(UOFFISIELL OVERSETTELSE)
") NOR/313R1031.fral COMMISSION IMPLEMENTING REGULATION (EU) No 1031/2013 of 24 October 2013 approving the active substance penflufen, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
NOR/313R1031.fral COMMISSION IMPLEMENTING REGULATION (EU) No 1031/2013 of 24 October 2013 approving the active substance penflufen, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
Forskrift om endringer i forskrift om begrensning i bruk av helse- og miljøfarlige kjemikalier og andre produkter (produktforskriften)
") Forskrift om endringer i forskrift om begrensning i bruk av helse- og miljøfarlige kjemikalier og andre produkter (produktforskriften) Hjemmel: Fastsatt av Klima- og miljødepartementet xx. xx. xxxx med
Forskrift om endringer i forskrift om begrensning i bruk av helse- og miljøfarlige kjemikalier og andre produkter (produktforskriften) Hjemmel: Fastsatt av Klima- og miljødepartementet xx. xx. xxxx med
Legemiddelpakke I nå også i Norge. Regulatorisk høstmøte Ingeniørenes hus, Oslo Onsdag 7.oktober 2009 Sidsel Harby Rådgiver, Statens legemiddelverk
 Legemiddelpakke I nå også i Norge Regulatorisk høstmøte Ingeniørenes hus, Oslo Onsdag 7.oktober 2009 Sidsel Harby Rådgiver, Statens legemiddelverk Agenda DCP Braille (blindeskrift) Usertesting (Lesbarhetstesting)
Legemiddelpakke I nå også i Norge Regulatorisk høstmøte Ingeniørenes hus, Oslo Onsdag 7.oktober 2009 Sidsel Harby Rådgiver, Statens legemiddelverk Agenda DCP Braille (blindeskrift) Usertesting (Lesbarhetstesting)
POWEL DATABEHANDLERAVTALE
 POWEL DATABEHANDLERAVTALE mellom Powel AS. Behandlingsansvarlig og «Navn på selskap». Databehandler Innhold 1 Formålet med Databehandleravtalen... 3 2 Definisjoner... 3 3 Formål med behandlingen... 3 4
POWEL DATABEHANDLERAVTALE mellom Powel AS. Behandlingsansvarlig og «Navn på selskap». Databehandler Innhold 1 Formålet med Databehandleravtalen... 3 2 Definisjoner... 3 3 Formål med behandlingen... 3 4
Forslag til fem forskrifter som gjennomfører EUs varedirektiver
 Notat 1 av 7 31.10.2016 16/9856 /MACE Utarbeidet av Deres dato Deres referanse Cecilie Magnussen, tlf. 33412500 Til Høringsinstansene Arkivkode Forslag til fem forskrifter som gjennomfører EUs varedirektiver
Notat 1 av 7 31.10.2016 16/9856 /MACE Utarbeidet av Deres dato Deres referanse Cecilie Magnussen, tlf. 33412500 Til Høringsinstansene Arkivkode Forslag til fem forskrifter som gjennomfører EUs varedirektiver
Re - sterilisering av medisinsk engangsutstyr? Margrete Drønen 23.03.2012
 Re - sterilisering av medisinsk engangsutstyr? Margrete Drønen 23.03.2012 Prinsippgrunnlag: Formelle krav som angår kvaliteten av sterilt medisinsk utstyr finnes i lov og forskrifter om medisinsk utstyr.
Re - sterilisering av medisinsk engangsutstyr? Margrete Drønen 23.03.2012 Prinsippgrunnlag: Formelle krav som angår kvaliteten av sterilt medisinsk utstyr finnes i lov og forskrifter om medisinsk utstyr.
INNHOLD VEDLEGG 1 VEDLEGG 2 VEDLEGG 3 VEDLEGG 4 VEDLEGG 5. 1 Fastsatt av Statens teleforvaltning 2. januar 1996
 1 Fastsatt av Statens teleforvaltning Forskrift om elektromagnetisk kompatibilitet (EMC) for teleutstyr. Fastsatt av Statens teleforvaltning 2.januar 1996 med hjemmel i lov 23.juni 1995 nr. 39 om telekommunikasjon
1 Fastsatt av Statens teleforvaltning Forskrift om elektromagnetisk kompatibilitet (EMC) for teleutstyr. Fastsatt av Statens teleforvaltning 2.januar 1996 med hjemmel i lov 23.juni 1995 nr. 39 om telekommunikasjon
Samtidig foreslås å oppheve forskrift nr. 309 om norsk ansvarlig organ for
 Notat Dato: 21.11.2012 Saksnr.: 12/7171 Høringsnotat - forslag om endringer i lov om merking av forbruksvarer og opphevelse av forskrift om norsk ansvarlig organ for EUs miljømerke, gebyrer, standardkontrakt
Notat Dato: 21.11.2012 Saksnr.: 12/7171 Høringsnotat - forslag om endringer i lov om merking av forbruksvarer og opphevelse av forskrift om norsk ansvarlig organ for EUs miljømerke, gebyrer, standardkontrakt
Reviderte direktiver hva betyr det for deres bedrift?
 Reviderte direktiver hva betyr det for deres bedrift? Silje Bertheussen Rådgiver/jurist Utredninger og Regelverk Gunnstein Åsmund Hæreid Senior Ingeniør/Fagkoordinator TKO Dagens regelverk Måleinstrumentdirektivet
Reviderte direktiver hva betyr det for deres bedrift? Silje Bertheussen Rådgiver/jurist Utredninger og Regelverk Gunnstein Åsmund Hæreid Senior Ingeniør/Fagkoordinator TKO Dagens regelverk Måleinstrumentdirektivet
NOR/314R0632.ohfo OJ L 175/14, p. 1-5
 1 NOR/314R0632.ohfo OJ L 175/14, p. 1-5 COMMISSION IMPLEMENTING REGULATION (EU) No 632/2014 of 13 May 2014 approving the active substance flubendiamide, in accordance with Regulation (EC) No 1107/2009
1 NOR/314R0632.ohfo OJ L 175/14, p. 1-5 COMMISSION IMPLEMENTING REGULATION (EU) No 632/2014 of 13 May 2014 approving the active substance flubendiamide, in accordance with Regulation (EC) No 1107/2009
(UOFFISIELL OVERSETTELSE)
") NOR/311R0993.jaeg OJ L 263/11, p. 1-4 COMMISSION IMPLEMENTING REGULATION (EU) No 993/2011 of 6 October 2011 approving the active substance 8-hydroxyquinoline, in accordance with Regulation (EC) No 1107/2009
NOR/311R0993.jaeg OJ L 263/11, p. 1-4 COMMISSION IMPLEMENTING REGULATION (EU) No 993/2011 of 6 October 2011 approving the active substance 8-hydroxyquinoline, in accordance with Regulation (EC) No 1107/2009
Dokumentasjonskrav for byggevarer. MATHIEU VEULEMANS 16.09.2013, Virke Bygg og Anlegg, dokumentasjon i handel med byggevaren
 Dokumentasjonskrav for byggevarer MATHIEU VEULEMANS 16.09.2013, Virke Bygg og Anlegg, dokumentasjon i handel med byggevaren Hva skal jeg snakke om? 1. Hva slags dokumentasjon vi krever 2. Hva betyr CE-merking?
Dokumentasjonskrav for byggevarer MATHIEU VEULEMANS 16.09.2013, Virke Bygg og Anlegg, dokumentasjon i handel med byggevaren Hva skal jeg snakke om? 1. Hva slags dokumentasjon vi krever 2. Hva betyr CE-merking?
(UOFFISIELL OVERSETTELSE)
") NOR/313R0568.fral COMMISSION IMPLEMENTING REGULATION (EU) No 568/2013 of 18 June 2013 approving the active substance thymol, in accordance with Regulation (EC) No 1107/2009 of the European Parliament and
NOR/313R0568.fral COMMISSION IMPLEMENTING REGULATION (EU) No 568/2013 of 18 June 2013 approving the active substance thymol, in accordance with Regulation (EC) No 1107/2009 of the European Parliament and
NORSK LOVTIDEND Avd. I Lover og sentrale forskrifter mv. Utgitt i henhold til lov 19. juni 1969 nr. 53.
 NORSK LOVTIDEND Avd. I Lover og sentrale forskrifter mv. Utgitt i henhold til lov 19. juni 1969 nr. 53. Kunngjort 11. oktober 2017 kl. 13.50 PDF-versjon 17. oktober 2017 10.10.2017 nr. 1598 Forskrift om
NORSK LOVTIDEND Avd. I Lover og sentrale forskrifter mv. Utgitt i henhold til lov 19. juni 1969 nr. 53. Kunngjort 11. oktober 2017 kl. 13.50 PDF-versjon 17. oktober 2017 10.10.2017 nr. 1598 Forskrift om
(UOFFISIELL OVERSETTELSE)
") NOR/311R0736.jaeg OJ L 195/11, p. 37-41 COMMISSION IMPLEMENTING REGULATION (EU) No 736/2011 of 26 July 2011 approving the active substance fluroxypyr, in accordance with Regulation (EC) No 1107/2009 of
NOR/311R0736.jaeg OJ L 195/11, p. 37-41 COMMISSION IMPLEMENTING REGULATION (EU) No 736/2011 of 26 July 2011 approving the active substance fluroxypyr, in accordance with Regulation (EC) No 1107/2009 of
Produksjonskurs. 13. Januar 2015
 Produksjonskurs 13. Januar 2015 CE merking av takstoler Myndighetskrav og regler Hvem er sertifisert? 53 bedrifter som produserer takstoler 49 bedrifter som produserer k-virke Hva betyr CE-merking? CE
Produksjonskurs 13. Januar 2015 CE merking av takstoler Myndighetskrav og regler Hvem er sertifisert? 53 bedrifter som produserer takstoler 49 bedrifter som produserer k-virke Hva betyr CE-merking? CE
Nedenfor gjengis til informasjon EØS-avtalen vedlegg I kapittel I del 7.2 (forordning (EF) nr. 878/2004) slik Mattilsynet tolker denne del av
 nr. 878/2004) slik Mattilsynet tolker denne del av") Nedenfor gjengis til informasjon EØS-avtalen vedlegg I kapittel I del 7.2 (forordning (EF) nr. 878/2004) slik Mattilsynet tolker denne del av EØS-avtalen med de endringer og tillegg som følger av EØS-tilpasningen
Nedenfor gjengis til informasjon EØS-avtalen vedlegg I kapittel I del 7.2 (forordning (EF) nr. 878/2004) slik Mattilsynet tolker denne del av EØS-avtalen med de endringer og tillegg som følger av EØS-tilpasningen
(UOFFISIELL OVERSETTELSE)
") NOR/313R0829.fral COMMISSION IMPLEMENTING REGULATION (EU) No 829/2013 of 29 August 2013 approving the active substance Pseudomonas sp. strain DSMZ 13134, in accordance with Regulation (EC) No 1107/2009
NOR/313R0829.fral COMMISSION IMPLEMENTING REGULATION (EU) No 829/2013 of 29 August 2013 approving the active substance Pseudomonas sp. strain DSMZ 13134, in accordance with Regulation (EC) No 1107/2009
NOR/304R T OJ L 379/05, p
 NOR/304R2230.00T OJ L 379/05, p. 64-67 Commission Regulation (EC) No 2230/2004 of 23 December 2004 laying down detailed rules for the implementation of European Parliament and Council Regulation (EC) No
NOR/304R2230.00T OJ L 379/05, p. 64-67 Commission Regulation (EC) No 2230/2004 of 23 December 2004 laying down detailed rules for the implementation of European Parliament and Council Regulation (EC) No
Høring - forslag til ny forskrift om håndtering av medisinsk utstyr
 Til: I følge adresseliste Høring - forslag til ny forskrift om håndtering av medisinsk utstyr 1. Innledning Helsedirektoratet og Direktoratet for samfunnssikkerhet og beredskap (DSB) fikk henholdsvis i
Til: I følge adresseliste Høring - forslag til ny forskrift om håndtering av medisinsk utstyr 1. Innledning Helsedirektoratet og Direktoratet for samfunnssikkerhet og beredskap (DSB) fikk henholdsvis i
Fakta September 2013 SIKKERHETSKRAV TIL LEKETØY. Barn skal ha den best tenkelige beskyttelse SIKKERHETSKRAV TIL LEKETØY
 Fakta September 2013 SIKKERHETSKRAV TIL LEKETØY SIKKERHETSKRAV TIL LEKETØY Barn skal ha den best tenkelige beskyttelse Det finnes ca. 80 millioner barn under 14 år i EU/EØS og rundt 2 000 virksomheter
Fakta September 2013 SIKKERHETSKRAV TIL LEKETØY SIKKERHETSKRAV TIL LEKETØY Barn skal ha den best tenkelige beskyttelse Det finnes ca. 80 millioner barn under 14 år i EU/EØS og rundt 2 000 virksomheter
Litt om Direktoratet for byggkvalitet
 Utpeking av TKO for produkter til byggverk MATHIEU VEULEMANS 08.09.2014, Akkrediteringsdagen, Park inn Oslo Airport Hotel Litt om Direktoratet for byggkvalitet 1 Direktoratet for byggkvalitet Avdeling
Utpeking av TKO for produkter til byggverk MATHIEU VEULEMANS 08.09.2014, Akkrediteringsdagen, Park inn Oslo Airport Hotel Litt om Direktoratet for byggkvalitet 1 Direktoratet for byggkvalitet Avdeling
Nr. 8/244 EØS-tillegget til Den europeiske unions tidende. KOMMISJONSFORORDNING (EU) nr. 143/2011
 nr. 143/2011") Nr. 8/244 EØS-tillegget til Den europeiske unions tidende KOMMISJONSFORORDNING (EU) nr. 143/2011 2016/EØS/8/42 av 17. februar 2011 om endring av vedlegg XIV til europaparlaments- og rådsforordning (EF)
Nr. 8/244 EØS-tillegget til Den europeiske unions tidende KOMMISJONSFORORDNING (EU) nr. 143/2011 2016/EØS/8/42 av 17. februar 2011 om endring av vedlegg XIV til europaparlaments- og rådsforordning (EF)
Forskrift om kvalitet på melk og melkeprodukter
 Forskrift om kvalitet på melk og melkeprodukter Veiledning om - opplysninger om faktisk fettinnhold som en del av betegnelsen på melk og melkeprodukter - forholdet til bestemmelsene i matinformasjonsforskriften
Forskrift om kvalitet på melk og melkeprodukter Veiledning om - opplysninger om faktisk fettinnhold som en del av betegnelsen på melk og melkeprodukter - forholdet til bestemmelsene i matinformasjonsforskriften
KOMMISJONENS DELEGERTE FORORDNING (EU) Nr. 574/2014. av 21. februar 2014
 Nr. 574/2014. av 21. februar 2014") KOMMISJONENS DELEGERTE FORORDNING (EU) Nr. 574/2014 av 21. februar 2014 om endring av vedlegg III til Europaparlamentets og Rådets forordning (EU) nr. 305/2011 hva angår malen som skal benyttes for utarbeidelse
KOMMISJONENS DELEGERTE FORORDNING (EU) Nr. 574/2014 av 21. februar 2014 om endring av vedlegg III til Europaparlamentets og Rådets forordning (EU) nr. 305/2011 hva angår malen som skal benyttes for utarbeidelse
HØRING - INNFØRING AV OVERTREDELSESGEBYR M.V. I FLERE LOVER MED FOLKEHELSEFORMÅL M.V.
 Lab Norge Laboratorieleverandørene Helse- og omsorgsdepartementet Pb 8011 Dep 0030 OSLO Oslo 30.8.2017 HØRING - INNFØRING AV OVERTREDELSESGEBYR M.V. I FLERE LOVER MED FOLKEHELSEFORMÅL M.V. Vi viser til
Lab Norge Laboratorieleverandørene Helse- og omsorgsdepartementet Pb 8011 Dep 0030 OSLO Oslo 30.8.2017 HØRING - INNFØRING AV OVERTREDELSESGEBYR M.V. I FLERE LOVER MED FOLKEHELSEFORMÅL M.V. Vi viser til
(UOFFISIELL OVERSETTELSE)
") NOR/313R0570.fral COMMISSION IMPLEMENTING REGULATION (EU) No 570/2013 of 17 June 2013 approving the active substance geraniol, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
NOR/313R0570.fral COMMISSION IMPLEMENTING REGULATION (EU) No 570/2013 of 17 June 2013 approving the active substance geraniol, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
(UOFFISIELL OVERSETTELSE)
") NOR/313R0827.fral COMMISSION IMPLEMENTING REGULATION (EU) No 827/2013 of 29 August 2013 approving the active substance Aureobasidium pullulans (strains DSM 14940 and DSM 14941), in accordance with Regulation
NOR/313R0827.fral COMMISSION IMPLEMENTING REGULATION (EU) No 827/2013 of 29 August 2013 approving the active substance Aureobasidium pullulans (strains DSM 14940 and DSM 14941), in accordance with Regulation
FÅ KONTROLL PÅ DE USTRUKTURERTE DATAENE
 FÅ KONTROLL PÅ DE USTRUKTURERTE DATAENE Start din reise mot å etterleve de nye personvernreglene INTRODUKSJON I mai 2018 innføres ny personvernlovgivning i Norge. Disse har vært mye omtalt, både som de
FÅ KONTROLL PÅ DE USTRUKTURERTE DATAENE Start din reise mot å etterleve de nye personvernreglene INTRODUKSJON I mai 2018 innføres ny personvernlovgivning i Norge. Disse har vært mye omtalt, både som de
Nærings- og fiskeridepartementet Postboks 8090 Dep 0033 OSLO
 W Nærings- og fiskeridepartementet Postboks 8090 Dep 0033 OSLO Vår ref.:1506244-2 - 414.2 Vår dato: 29.2.2016 Deres ref.: 15/4361 Deres dato: 30.11.2015 Saksbehandler: Kristina Mari Rognmo Svar på høring
W Nærings- og fiskeridepartementet Postboks 8090 Dep 0033 OSLO Vår ref.:1506244-2 - 414.2 Vår dato: 29.2.2016 Deres ref.: 15/4361 Deres dato: 30.11.2015 Saksbehandler: Kristina Mari Rognmo Svar på høring
REGULATION (EU) 2017/746 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive
 2017/746 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive") REGULATION (EU) 2017/746 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU 1 2 EUROPAPARLAMENTS-
REGULATION (EU) 2017/746 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU 1 2 EUROPAPARLAMENTS-
(UOFFISIELL OVERSETTELSE)
") NOR/313R1177.fral COMMISSION IMPLEMENTING REGULATION (EU) No 1177/2013 of 20 November 2013 approving the active substance spirotetramat, in accordance with Regulation (EC) No 1107/2009 of the European
NOR/313R1177.fral COMMISSION IMPLEMENTING REGULATION (EU) No 1177/2013 of 20 November 2013 approving the active substance spirotetramat, in accordance with Regulation (EC) No 1107/2009 of the European
(UOFFISIELL OVERSETTELSE)
") NOR/313R0208.fral OJ L 68 /13, p. 16-18 COMMISSION IMPLEMENTING REGULATION (EU) No 208/2013 of 11 March 2013 on traceability requirements for sprouts and seeds intended for the production of sprouts (UOFFISIELL
NOR/313R0208.fral OJ L 68 /13, p. 16-18 COMMISSION IMPLEMENTING REGULATION (EU) No 208/2013 of 11 March 2013 on traceability requirements for sprouts and seeds intended for the production of sprouts (UOFFISIELL
(UOFFISIELL OVERSETTELSE)
") NOR/313R1187.fral COMMISSION IMPLEMENTING REGULATION (EU) No 1187/2013 of 21 November 2013 approving the active substance penthiopyrad, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
NOR/313R1187.fral COMMISSION IMPLEMENTING REGULATION (EU) No 1187/2013 of 21 November 2013 approving the active substance penthiopyrad, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
Veileder for anskaffelse av datamodem
 TEMA Veileder for anskaffelse av datamodem Produkter og tjenester i Nødnett Mai 2017 Utgitt av: Direktoratet for samfunnssikkerhet og beredskap (DSB) 2017 ISBN: Omslagsfoto: Grafisk produksjon: 978-82-7768-435-2
TEMA Veileder for anskaffelse av datamodem Produkter og tjenester i Nødnett Mai 2017 Utgitt av: Direktoratet for samfunnssikkerhet og beredskap (DSB) 2017 ISBN: Omslagsfoto: Grafisk produksjon: 978-82-7768-435-2
(UOFFISIELL OVERSETTELSE)
") NOR/313R0188.fral COMMISSION IMPLEMENTING REGULATION (EU) No 188/2013 of 5 March 2013 approving the active substance mandipropamid, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
NOR/313R0188.fral COMMISSION IMPLEMENTING REGULATION (EU) No 188/2013 of 5 March 2013 approving the active substance mandipropamid, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
COMMISSION IMPLEMENTING REGULATION (EU) 2016/9 of 5 January 2016 on joint submission of data and datasharing in accordance with Regulation (EC) No
 2016/9 of 5 January 2016 on joint submission of data and datasharing in accordance with Regulation (EC) No") COMMISSION IMPLEMENTING REGULATION (EU) 2016/9 of 5 January 2016 on joint submission of data and datasharing in accordance with Regulation (EC) No 1907/2006 of the European Parliament and of the Council
COMMISSION IMPLEMENTING REGULATION (EU) 2016/9 of 5 January 2016 on joint submission of data and datasharing in accordance with Regulation (EC) No 1907/2006 of the European Parliament and of the Council
(UOFFISIELL OVERSETTELSE)
") NOR/313R1175.fral COMMISSION IMPLEMENTING REGULATION (EU) No 1175/2013 of 20 November 2013 approving the active substance benalaxyl-m, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
NOR/313R1175.fral COMMISSION IMPLEMENTING REGULATION (EU) No 1175/2013 of 20 November 2013 approving the active substance benalaxyl-m, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
(UOFFISIELL OVERSETTELSE)
") NOR/313R0613.fral OJ L 173/13, p. 34-37 COMMISSION REGULATION (EU) No 613/2013 of 25 June 2013 amending Regulation (EC) No 1451/2007 as regards additional active substances of biocidal products to be examined
NOR/313R0613.fral OJ L 173/13, p. 34-37 COMMISSION REGULATION (EU) No 613/2013 of 25 June 2013 amending Regulation (EC) No 1451/2007 as regards additional active substances of biocidal products to be examined
Direktoratet for samfunnssikkerhet og beredskap
 Direktoratet for samfunnssikkerhet og beredskap DSBs Myndighetsområde for Elektrisk utstyr Seksjon for produktsikkerhet (PRO) Bjørn Nyrud 18. oktober 2018 Visjon og mål Alt elektrisk utstyr i Norge skal
Direktoratet for samfunnssikkerhet og beredskap DSBs Myndighetsområde for Elektrisk utstyr Seksjon for produktsikkerhet (PRO) Bjørn Nyrud 18. oktober 2018 Visjon og mål Alt elektrisk utstyr i Norge skal
(UOFFISIELL OVERSETTELSE)
") NOR/312R0595.fral COMMISSION IMPLEMENTING REGULATION (EU) No 595/2012 of 5 July 2012 approving the active substance fenpyrazamine, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
NOR/312R0595.fral COMMISSION IMPLEMENTING REGULATION (EU) No 595/2012 of 5 July 2012 approving the active substance fenpyrazamine, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
Kontakt- og informasjonsmøte om produktdokumentasjon SINTEF Byggforsk. 6.mars Byggevareforordningen Konsekvenser for byggenæringen
 Kontakt- og informasjonsmøte om produktdokumentasjon SINTEF Byggforsk 6.mars 2012 Byggevareforordningen Konsekvenser for byggenæringen Arne Skjelle Byggenæringens Landsforening Construction Products Regulation
Kontakt- og informasjonsmøte om produktdokumentasjon SINTEF Byggforsk 6.mars 2012 Byggevareforordningen Konsekvenser for byggenæringen Arne Skjelle Byggenæringens Landsforening Construction Products Regulation
Høring om forslag til endringer i forskrift 21.desember 2007 nr om krav til ikke-automatiske vekter og andre instrumentspesifikke forskrifter
 Høring om forslag til endringer i forskrift 21.desember 2007 nr. 1735 om krav til ikke-automatiske vekter og andre instrumentspesifikke forskrifter 1 Hovedinnholdet i forslaget Følgende forskrifter sendes
Høring om forslag til endringer i forskrift 21.desember 2007 nr. 1735 om krav til ikke-automatiske vekter og andre instrumentspesifikke forskrifter 1 Hovedinnholdet i forslaget Følgende forskrifter sendes
NOR/306R T OJ L 360/2006, p
 NOR/306R1877.00T OJ L 360/2006, p. 133-136 COMMISSION REGULATION (EC) No 1877/2006 of 18 December 2006 amending Regulation (EC) No 878/2004 laying down transitional measures in accordance with Regulation
NOR/306R1877.00T OJ L 360/2006, p. 133-136 COMMISSION REGULATION (EC) No 1877/2006 of 18 December 2006 amending Regulation (EC) No 878/2004 laying down transitional measures in accordance with Regulation
Nedenfor gjengis til informasjon EØS-avtalen vedlegg I kapittel I del 7.2 (forordning (EF) nr. 181/2006) slik Mattilsynet tolker denne del av
 nr. 181/2006) slik Mattilsynet tolker denne del av") Nedenfor gjengis til informasjon EØS-avtalen vedlegg I kapittel I del 7.2 (forordning (EF) nr. 181/2006) slik Mattilsynet tolker denne del av EØS-avtalen med de endringer og tillegg som følger av EØS-tilpasningen
Nedenfor gjengis til informasjon EØS-avtalen vedlegg I kapittel I del 7.2 (forordning (EF) nr. 181/2006) slik Mattilsynet tolker denne del av EØS-avtalen med de endringer og tillegg som følger av EØS-tilpasningen
(UOFFISIELL OVERSETTELSE)
") NOR/314R0144.beja OJ L 45/14, p. 7-11 COMMISSION IMPLEMENTING REGULATION (EU) No 144/2014 of 14 February 2014 approving the active substance valifenalate, in accordance with Regulation (EC) No 1107/2009
NOR/314R0144.beja OJ L 45/14, p. 7-11 COMMISSION IMPLEMENTING REGULATION (EU) No 144/2014 of 14 February 2014 approving the active substance valifenalate, in accordance with Regulation (EC) No 1107/2009
NOR/305R T OJ L 329/05, p. 4-7
 NOR/305R2049.00T OJ L 329/05, p. 4-7 COMMISSION REGULATION (EC) No 2049/2005 of 15 December 2005 laying down, pursuant to Regulation (EC) No 726/2004 of the European Parliament and of the Council, rules
NOR/305R2049.00T OJ L 329/05, p. 4-7 COMMISSION REGULATION (EC) No 2049/2005 of 15 December 2005 laying down, pursuant to Regulation (EC) No 726/2004 of the European Parliament and of the Council, rules
Høringsnotat. Innhold. 1. Innledning
 Høringsnotat Innhold 1. Innledning... 1 2. Kommisjonens gjennomføringsforordning (EU) 2015/806 av 22. mai 2015 med spesifikasjoner om formatet til EU-tillitsmerke for kvalifiserte tillitstjenester... 3
Høringsnotat Innhold 1. Innledning... 1 2. Kommisjonens gjennomføringsforordning (EU) 2015/806 av 22. mai 2015 med spesifikasjoner om formatet til EU-tillitsmerke for kvalifiserte tillitstjenester... 3
(UOFFISIELL OVERSETTELSE)
") NOR/313R0826.fral COMMISSION IMPLEMENTING REGULATION (EU) No 826/2013 of 29 August 2013 approving the active substance sedaxane, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
NOR/313R0826.fral COMMISSION IMPLEMENTING REGULATION (EU) No 826/2013 of 29 August 2013 approving the active substance sedaxane, in accordance with Regulation (EC) No 1107/2009 of the European Parliament
COMMISSION IMPLEMENTING REGULATION (EU) 2016/370 of 15 March 2016 approving the active substance pinoxaden, in accordance with Regulation (EC) No
 2016/370 of 15 March 2016 approving the active substance pinoxaden, in accordance with Regulation (EC) No") COMMISSION IMPLEMENTING REGULATION (EU) 2016/370 of 15 March 2016 approving the active substance pinoxaden, in accordance with Regulation (EC) No 1107/2009 of the European Parliament and of the Council
COMMISSION IMPLEMENTING REGULATION (EU) 2016/370 of 15 March 2016 approving the active substance pinoxaden, in accordance with Regulation (EC) No 1107/2009 of the European Parliament and of the Council
(UOFFISIELL OVERSETTELSE)
") NOR/313R0348.tona OJ L 108/13, p. 1-5 COMMISSION REGULATION (EU) No 348/2013 of 17 April 2013 amending Annex XIV to Regulation (EC) No 1907/2006 of the European Parliament and of the Council on the Registration,
NOR/313R0348.tona OJ L 108/13, p. 1-5 COMMISSION REGULATION (EU) No 348/2013 of 17 April 2013 amending Annex XIV to Regulation (EC) No 1907/2006 of the European Parliament and of the Council on the Registration,